RINVOQ is an oral selective and reversible JAK inhibitor indicated for the treatment of active ankylosing spondylitis in adult patients who have responded inadequately to conventional therapy.1




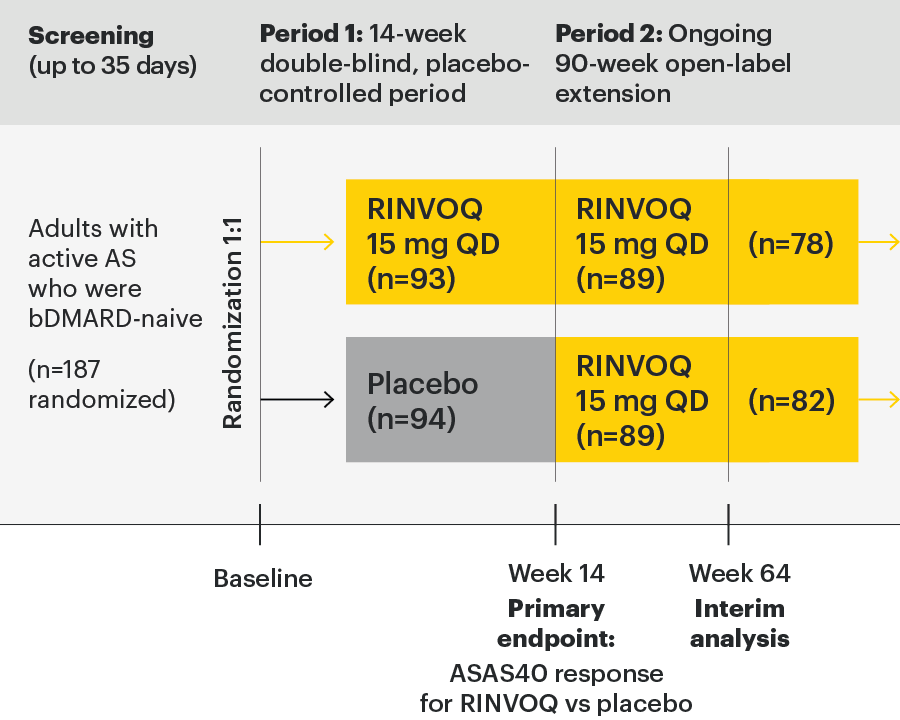
Permitted concomitant medications were csDMARDs (methotrexate, leflunomide, sulfasalazine, or hydroxychloroquine), oral glucocorticoids, NSAIDs, and analgesics.
ASAS40: at least 40% improvement in Assessment of Ankylosing Spondylitis International Working Group criteria; BASDAI: Bath Ankylosing Spondylitis Disease Activity Index; bDMARD: biologic disease-modifying antirheumatic drug; csDMARD: conventional synthetic disease-modifying antirheumatic drug; JAK: Janus kinase; NSAID: nonsteroidal anti-inflammatory drug; QD: once daily.
Primary
ASAS40 response for RINVOQ vs placebo at Week 14
Secondary (RINVOQ vs placebo at Week 14)*
• Change from baseline ASDAS†
• Change from baseline in SPARCC-MRI spine score†
• Proportion of patients achieving BASDAl50†
• Proportion of patients achieving ASAS partial remission†
• Change from baseline in BASFI†
• Change from baseline in ASQoL‡
• Change from baseline in BASMI‡
• Change from baseline in MASES‡
• Change from baseline in WPAI§
• Change from baseline in ASAS health index‡
Safety Assessments
Data for treatment-emergent adverse events and laboratory assessments were collected during the study. Treatment-emergent adverse events were defined as adverse events that began or worsened in severity after the first dose of study medication through 30 days after the last dose.
*The multiplicity-controlled secondary endpoints were tested in a sequential manner: ASDAS, SPARCC-MRI spine, group of endpoints tested by Hochberg procedure (BASDAl 50, ASQoL, ASAS partial remission, BASFI, BASMI, MASES, and WPAI), and ASAS Health Index. ASAS Health Index could not be evaluated within the multiplicity-controlled sequence because some endpoints in the Hochberg procedure were not significant (NS).
†Statistically significant in multiplicity-controlled analysis.
‡Not multiplicity-controlled; nominal P<0.05 vs placebo. No clinical inferences can be drawn.
§Not multiplicity-controlled. No clinical inferences can be drawn.
ASAS: Assessment of Ankylosing Spondylitis International Society; ASDAS: Ankylosing Spondylitis Disease Activity Score; ASQoL: ankylosing spondylitis quality of life; BASDAI 50: at least 50% improvement in Bath Ankylosing Spondylitis Disease Activity Index; BASFI: Bath Ankylosing Spondylitis Functional Index; BASMI: Bath Ankylosing Spondylitis Metrology Index; MASES: Maastricht Ankylosing Spondylitis Enthesitis Score; SPARCC MRI: Spondyloarthritis Research Consortium of Canada magnetic resonance imaging; WPAI: Work Productivity and Activity Impairment.
- Patients ≥18 years of age were eligible to participate
- Met the modified New York criteria for ankylosing spondylitis based on central reading of radiographs of the sacroiliac joints
- BASDAI score ≥4 at baseline
- Patient assessed back pain score ≥4 at screening and baseline
- Had inadequate response to at least 2 NSAIDs or intolerance to or contraindication for NSAIDs
- Patients with previous exposure to any JAK therapy or any biologic therapy with a potential effect on spondyloarthritis were excluded
- Patients with total spinal ankylosis were excluded from the study
Most patients were men (132 [71%]), were HLA-B27 positive (143 [76%]), and were receiving concomitant nonsteroidal anti-inflammatory drugs at baseline (152 [81%]). Baseline disease characteristics were generally balanced between the two groups.

EXPERT PERSPECTIVES

Watch an overview of the study design and results.2

NOTE TO AFFILIATES: Please evaluate y-axis scale throughout, per local regulations.
In patients with active AS and an inadequate response to conventional therapy
Significant improvement in AS signs and symptoms
SELECT-AXIS 1: ASAS40 at Week 14 (NRI)1,2
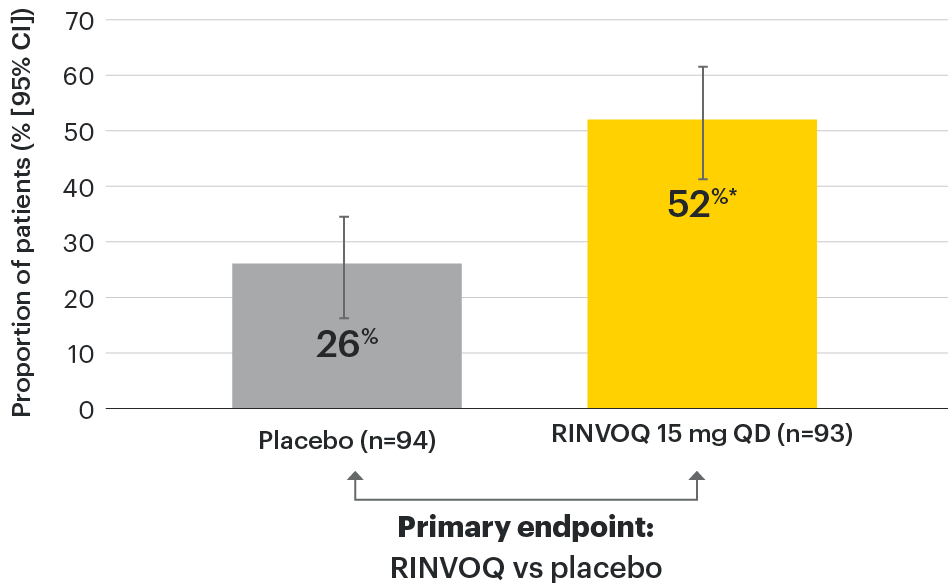

*P≤0.001 vs placebo, statistically significant in the multiplicity-controlled analysis.
95% confidence intervals are displayed as error bars in the chart, where available. Missing data were handled using NRI.
ASAS40: at least 40% improvement in Assessment of Ankylosing Spondylitis International Working Group criteria; NRI: nonresponder imputation; QD: once daily.
In patients with active AS and an inadequate response to conventional therapy
RINVOQ maintained ASAS40 response through 64 weeks1
SELECT-AXIS 1: ASAS40 response rates over time to Week 64 (NRI)1-3
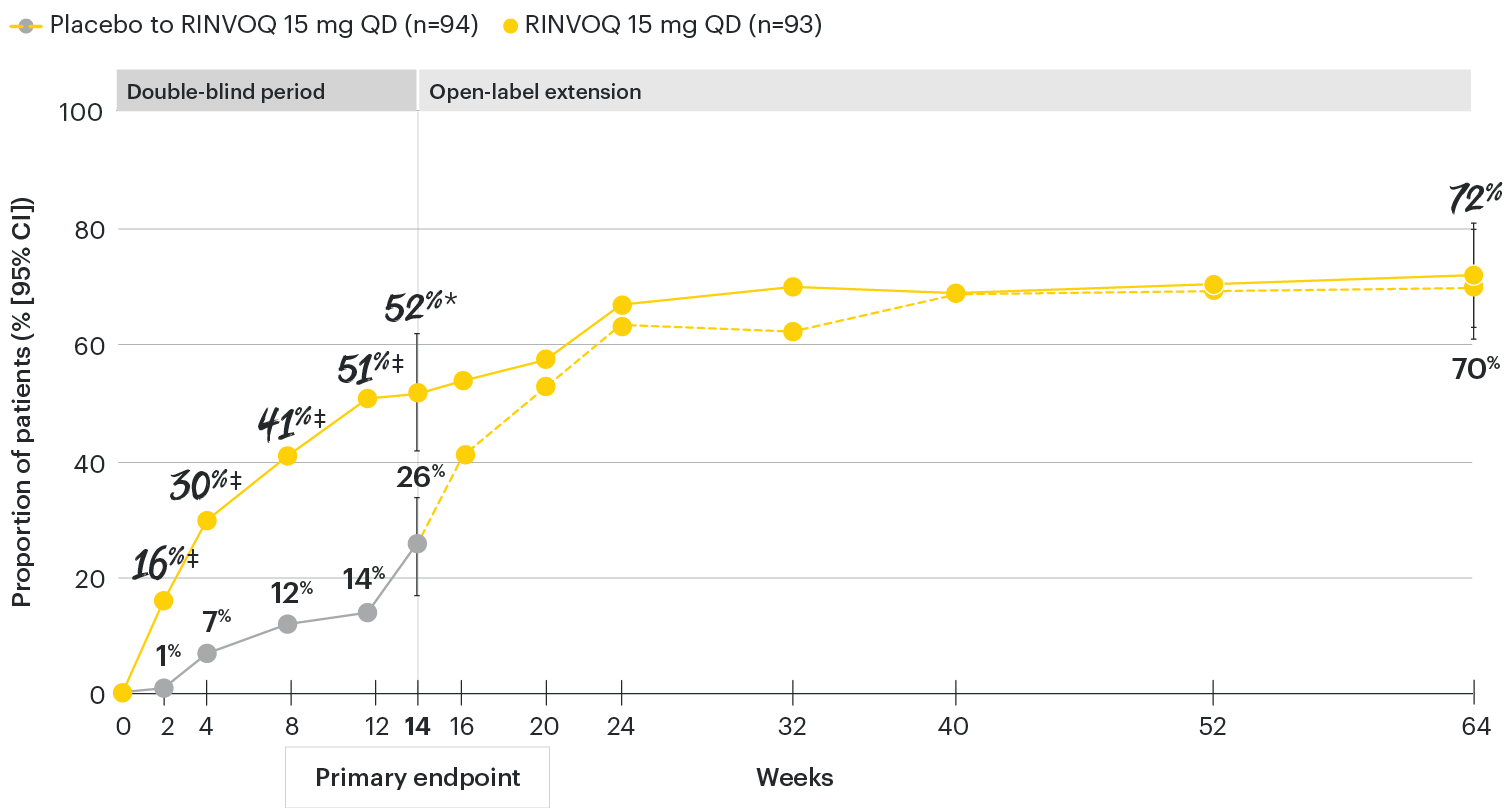
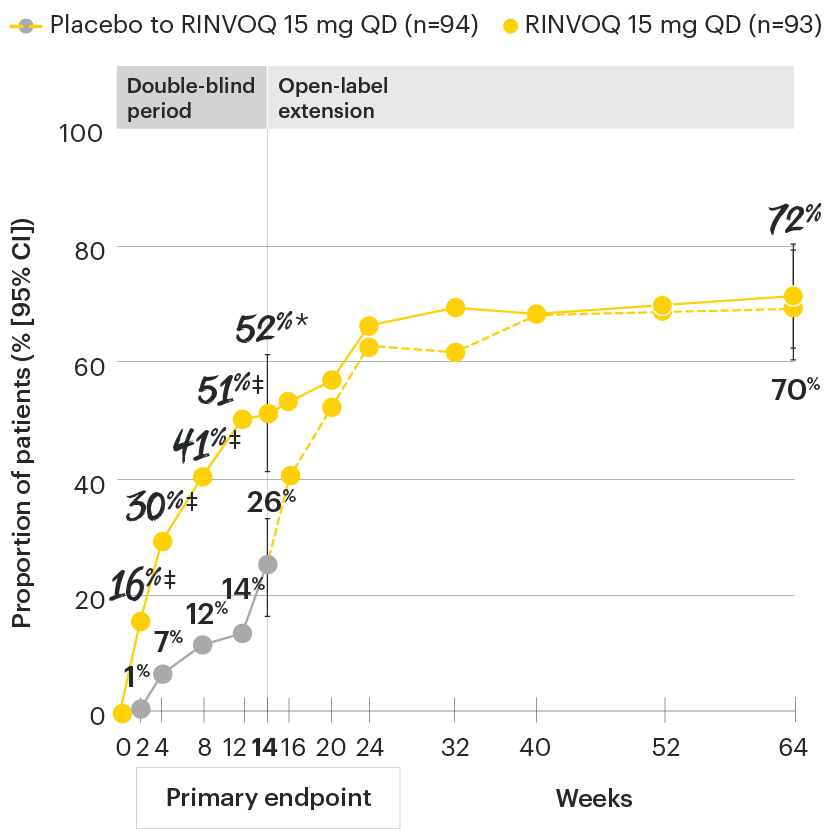
*P≤0.001 vs placebo, statistically significant in the multiplicity-controlled analysis.
‡Nominal P≤0.001 vs placebo, not multiplicity-controlled. No clinical inferences can be drawn.
DATA LIMITATIONS: Data not labeled as a primary or ranked secondary endpoint were prespecified, however they were not ranked or controlled for multiplicity; therefore, treatment differences could represent chance findings. No conclusions regarding these comparisons can be made.
ASAS40 with RINVOQ vs. placebo at Week 14 was a primary multiplicity-controlled endpoint, all other data shown were prespecified nonranked non–multiplicity-controlled endpoints. All patients randomized to placebo received open-label RINVOQ 15 mg QD starting from Week 14. Data shown through Week 14 are from the Week 14 data cut of SELECT-AXIS 1. Data through Week 64 are from the Week 64 data cut of SELECT-AXIS 1 and may include differences when compared with the primary Week 14 analysis.
95% confidence intervals are displayed as error bars in the chart, where available. Missing data were handled using NRI.
ASAS40: at least 40% improvement in Assessment of Ankylosing Spondylitis International Working Group criteria; NRI: nonresponder imputation; QD: once daily.

EXPERT PERSPECTIVES

Listen to Dr. Walter P. Maksymowych discuss RINVOQ study results in bDMARD-naive patients with active anklyosing spondylitis.1,2
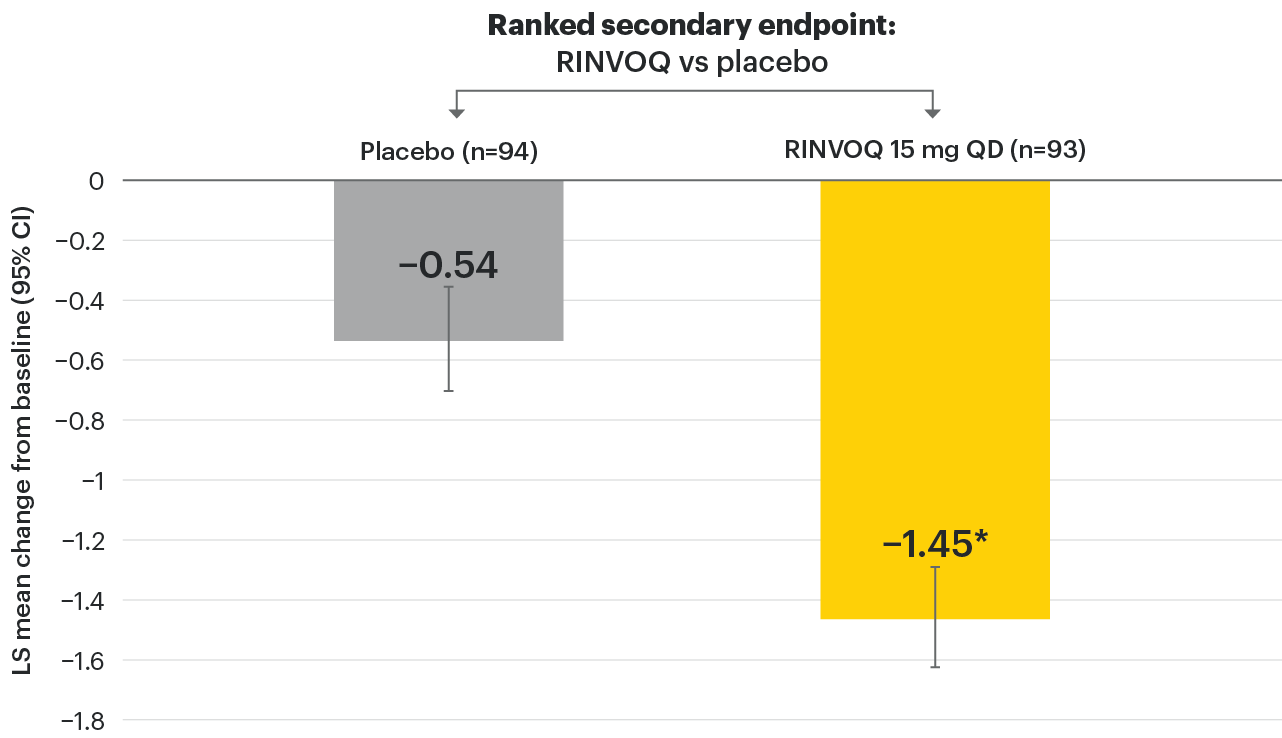
In patients with active AS and an inadequate response to conventional therapy
RINVOQ demonstrated a statistically significant change from baseline to Week 14 in ASDAS-CRP vs placebo
SELECT-AXIS 1: LS mean change in ASDAS-CRP from baseline to Week 14 (MMRM)1
ASDAS-CRP measures:2
- Spinal pain (back, neck, hips)
- Duration of morning stiffness
- Peripheral joint pain/swelling
- Patient global assessment of disease activity
- hs-CRP
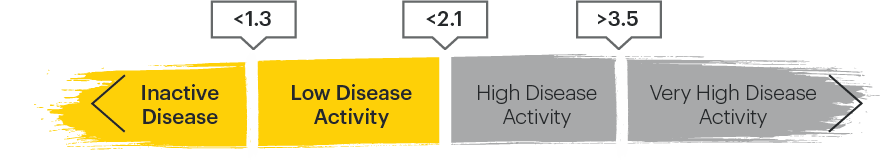
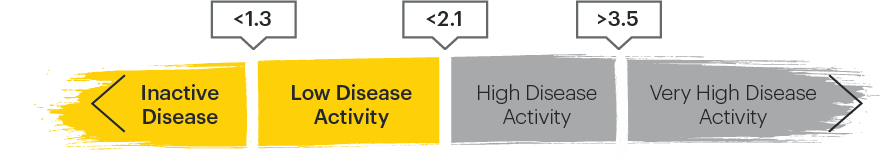
ASDAS categories2,4
Mean baseline ASDAS-CRP score:2
RINVOQ patients: 3.5
Placebo patients: 3.7
*P≤0.001 vs placebo, statistically significant in multiplicity-controlled analysis.
95% confidence intervals are displayed as error bars in the chart. Missing data were handled using MMRM.
ASDAS-CRP: Ankylosing Spondylitis Disease Activity Score with C-reactive protein; CI: confidence interval; LS: least squares; MMRM: mixed-effect model for repeated measures; QD: once daily.
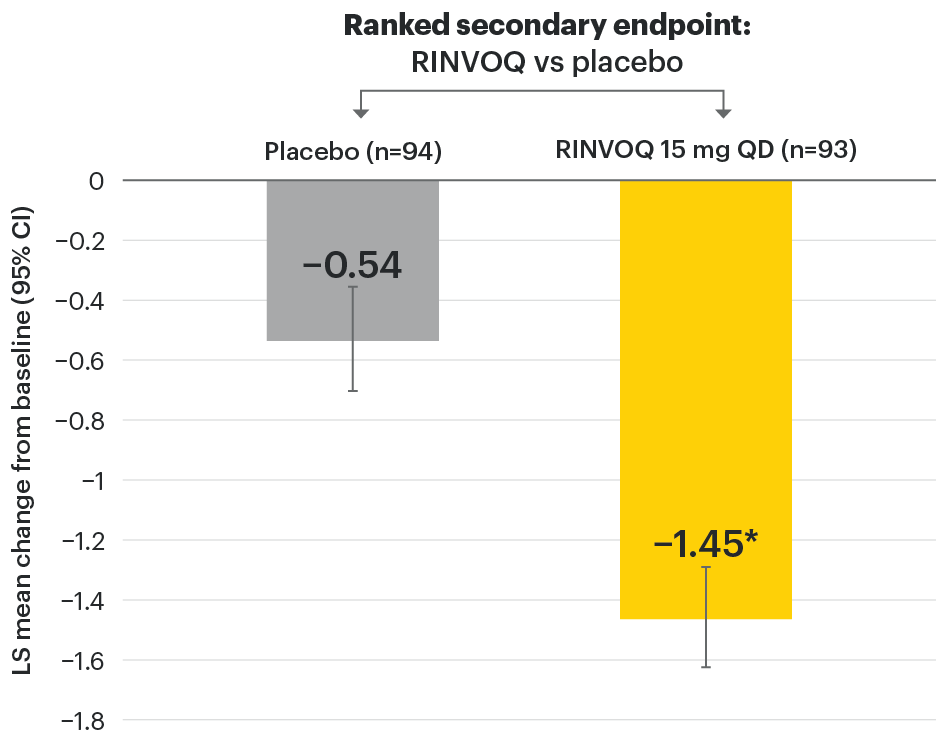
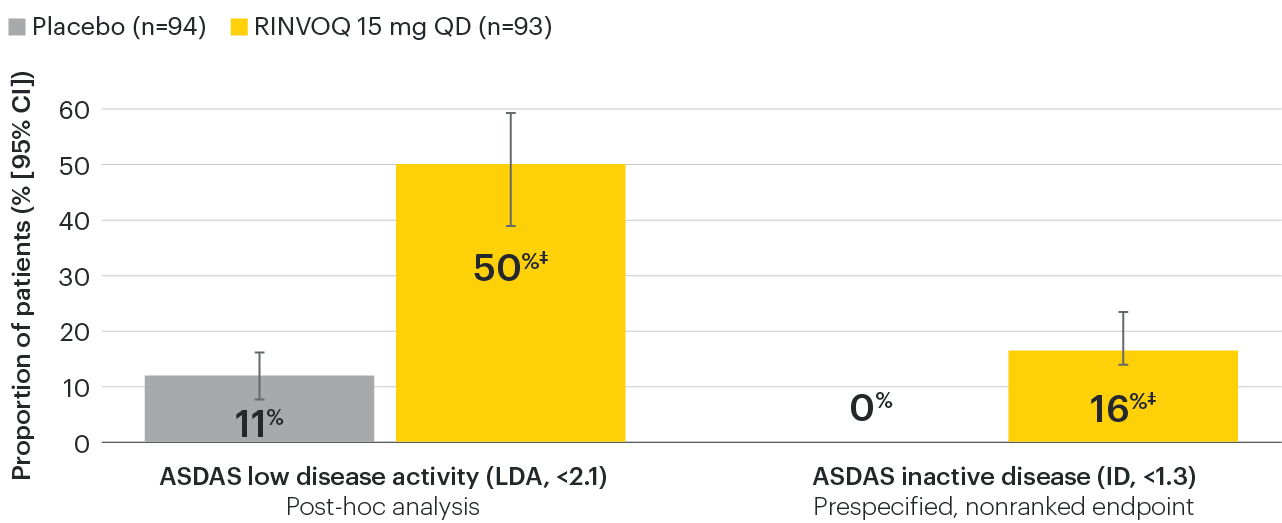
In patients with active AS and an inadequate response to conventional therapy
Disease control measures across ASDAS outcomes
SELECT-AXIS 1: ASDAS-LDA and ASDAS-ID response rates at Week 14 (NRI)1,2
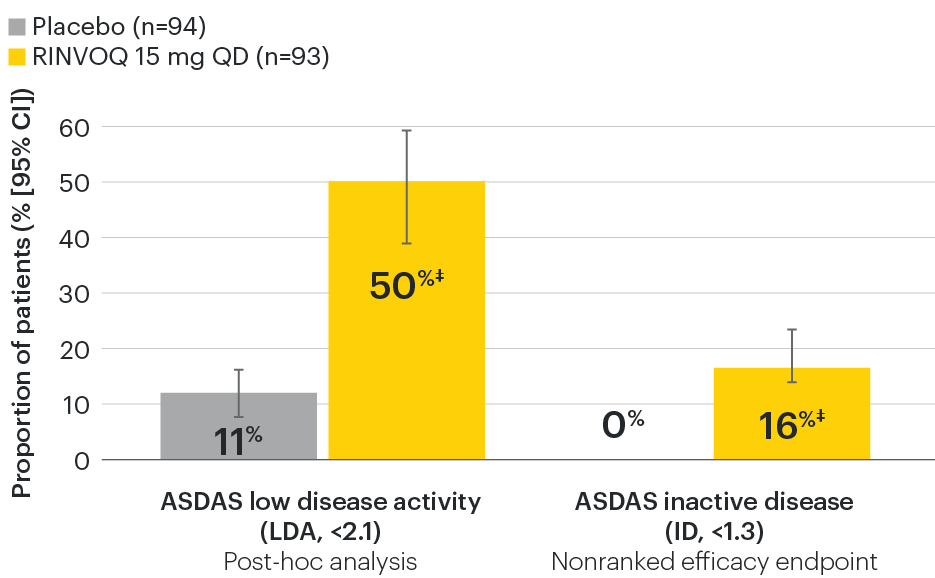
ASDAS-CRP measures:2
- Spinal pain (back, neck, hips)
- Duration of morning stiffness
- Peripheral joint pain/swelling
- Patient global assessment of disease activity
- hs-CRP
ASDAS categories2,4
Mean baseline ASDAS-CRP score:2
RINVOQ patients: 3.5
Placebo patients: 3.7
‡Nominal P≤0.001 vs placebo, not multiplicity-controlled. No clinical inferences can be drawn.
95% confidence intervals are displayed as error bars in the chart. Missing data were handled using NRI.
ASDAS: Ankylosing Spondylitis Disease Activity Index; ASDAS-ID: Ankylosing Spondylitis Disease Activity inactive disease; ASDAS-LDA: Ankylosing Spondylitis Disease Activity low disease activity; NRI: nonresponder imputation; QD: once daily.
In patients with active AS and an inadequate response to conventional therapy
ASDAS-CRP change from baseline over time
SELECT-AXIS 1: LS mean change in ASDAS-CRP from baseline to Week 64 (MMRM)2,3
NOTE TO AFFILIATES: Please evaluate references and presentation of data to Week 64, according to local policy, codes and regulations.


At baseline, mean ASDAS-CRP scores were 3.7 for placebo and 3.5 for RINVOQ 15 mg QD.2
All patients randomized to placebo received open-label RINVOQ 15 mg QD starting from Week 14. Data shown are from the 64-week analysis of SELECT-AXIS 1 using MMRM for missing data. Because MMRM utilizes all available data to estimate a value for missing data, there may be small differences in the data out to Week 64 when compared with earlier analyses.
DATA LIMITATIONS: Data not labeled as a ranked primary or secondary endpoint were prespecified, however they were not ranked or controlled for multiplicity; therefore, treatment differences could represent chance findings. No conclusions regarding these comparisons can be made.
ASDAS-CRP: Ankylosing Spondylitis Disease Activity Score with C-reactive protein; CI: confidence interval; LS: least squares; MMRM: mixed-effect model for repeated measures; QD: once daily.
In patients with active AS and an inadequate response to conventional therapy
ASDAS low disease activity (LDA <2.1) rates over time
SELECT-AXIS 1: ASDAS-LDA (<2.1) rates through Week 64 (NRI) (post-hoc analysis)1-3
NOTE TO AFFILIATES: Please evaluate references and presentation of data to Week 64, according to local policy, codes and regulations.


‡Nominal P≤0.001 vs placebo, not multiplicity-controlled. No clinical inferences can be drawn.
All patients randomized to placebo received open-label RINVOQ 15 mg QD starting from Week 14. Data shown are from the 64-week analysis of SELECT-AXIS 1.
95% confidence intervals are displayed as error bars in the chart, where available. Missing data were handled using NRI.
DATA LIMITATIONS: Data not labeled as a ranked primary or secondary endpoint were prespecified, however they were not ranked or controlled for multiplicity; therefore, treatment differences could represent chance findings. No conclusions regarding these comparisons can be made.
ASDAS: Ankylosing Spondylitis Disease Activity Index; ASDAS-LDA: Ankylosing Spondylitis Disease Activity low disease activity; NRI: nonresponder imputation; QD: once daily.
In patients with active AS and an inadequate response to conventional therapy
ASDAS inactive disease (ID <1.3) rates over time
SELECT-AXIS 1: ASDAS-ID (<1.3) rates through Week 64 (NRI)1-3
NOTE TO AFFILIATES: Please evaluate references and presentation of data to Week 64, according to local policy, codes and regulations.


‡Nominal P≤0.001 vs placebo, not multiplicity-controlled. No clinical inferences can be drawn.
ASDAS ID at Week 14 was a prespecified, nonranked endpoint.
All patients randomized to placebo received open-label RINVOQ 15 mg QD starting from Week 14. Data shown are from the 64-week analysis of SELECT-AXIS 1.
95% confidence intervals are displayed as error bars in the chart, where available. Missing data were handled using NRI.
DATA LIMITATIONS: Data not labeled as a ranked primary or secondary endpoint were prespecified, however they were not ranked or controlled for multiplicity; therefore, treatment differences could represent chance findings. No conclusions regarding these comparisons can be made.
ASDAS: Ankylosing Spondylitis Disease Activity Index; ASDAS-ID: Ankylosing Spondylitis Disease Activity inactive disease; NRI: nonresponder imputation; QD: once daily.


NOTE TO AFFILIATES: Please evaluate references and presentation of data at Week 14 and to 64, according to local policy, codes and regulations.
In patients with active AS and an inadequate response to conventional therapy
Improvement in back pain, function, and inflammation
SELECT-AXIS 1: Improvement across ASAS components at Week 14 (MMRM)1,2
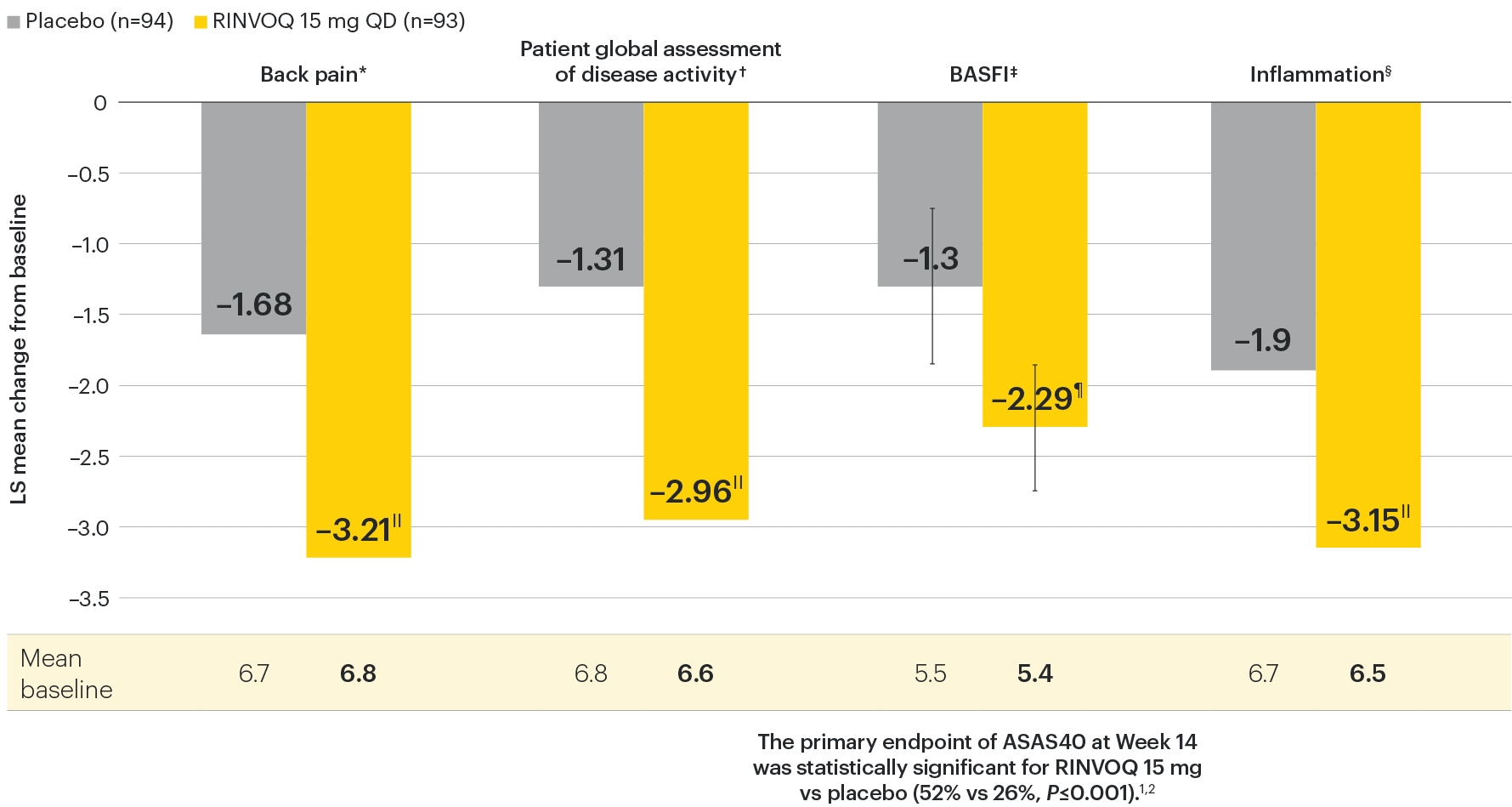
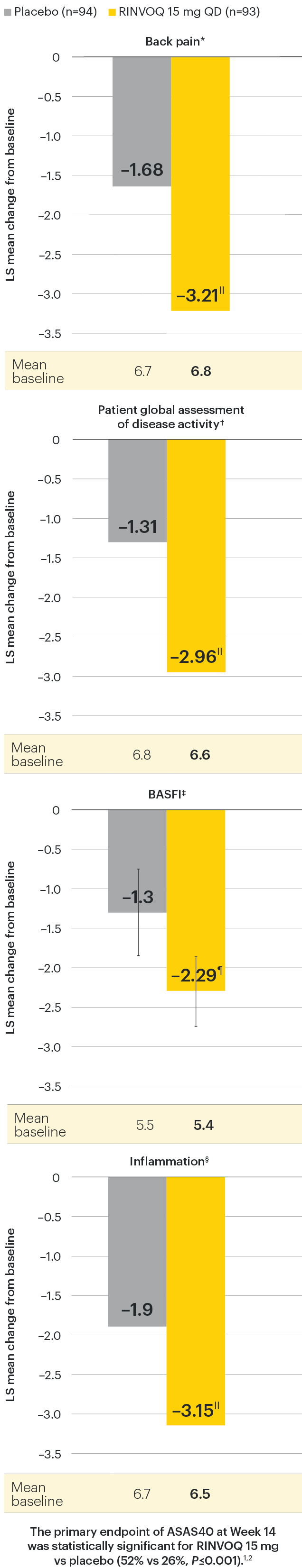
Change from baseline in BASFI for RINVOQ vs placebo at Week 14 was a ranked secondary endpoint in the Week 14 analysis of SELECT-AXIS 1.
IINominal P≤0.001 vs placebo, not multiplicity-controlled. No clinical inferences can be drawn.
¶P<0.01 vs placebo, statistically significant in multiplicity-controlled analysis.
DATA LIMITATIONS: Data not labeled as a ranked primary or secondary endpoint were prespecified, however they were not ranked or controlled for multiplicity; therefore, treatment differences could represent chance findings. No conclusions regarding these comparisons can be made.
*Back pain defined on a numerical rating scale (0–10) based on the following question: “What is the amount of back pain that you experienced at any time during the last week?”
†Patient's global assessment of disease activity was measured on a numerical rating scale ranging from 0 (no activity) to 10 (severe activity).5
‡Function was measured by the Bath Ankylosing Spondylitis Functional Index (BASFI), which consists of 10 items assessing participants' ability to perform activities on a numerical rating scale ranging from 0 (easy) to 10 (impossible).5
§Inflammation is represented by the mean of the two morning stiffness-related BASDAI NRS scores (mean of items 5 and 6 of the BASDAI [0–10]). BASDAI item 5 is based on the following question: “How would you describe the overall level of morning stiffness you have had from the time you wake up?” BASDAI item 6 is based on the following question: “How long does your morning stiffness last from the time you wake up?”
95% confidence intervals are displayed as error bars in the chart, where available. Missing data were handled using MMRM.
ASAS: Assessment of Ankylosing Spondylitis International Working Group criteria; BASDAI: Bath Ankylosing Spondylitis Disease Activity Index; BASFI: Bath Ankylosing Spondylitis Functional Index; CI: confidence interval; LS: least squares; MMRM: mixed-effect model for repeated measures; NRS: numeric rating scale.
In patients with active AS and an inadequate response to conventional therapy
Change in back pain from baseline over time
SELECT-AXIS 1: LS mean change in patient-reported back pain* from baseline to Week 64 (MMRM)3
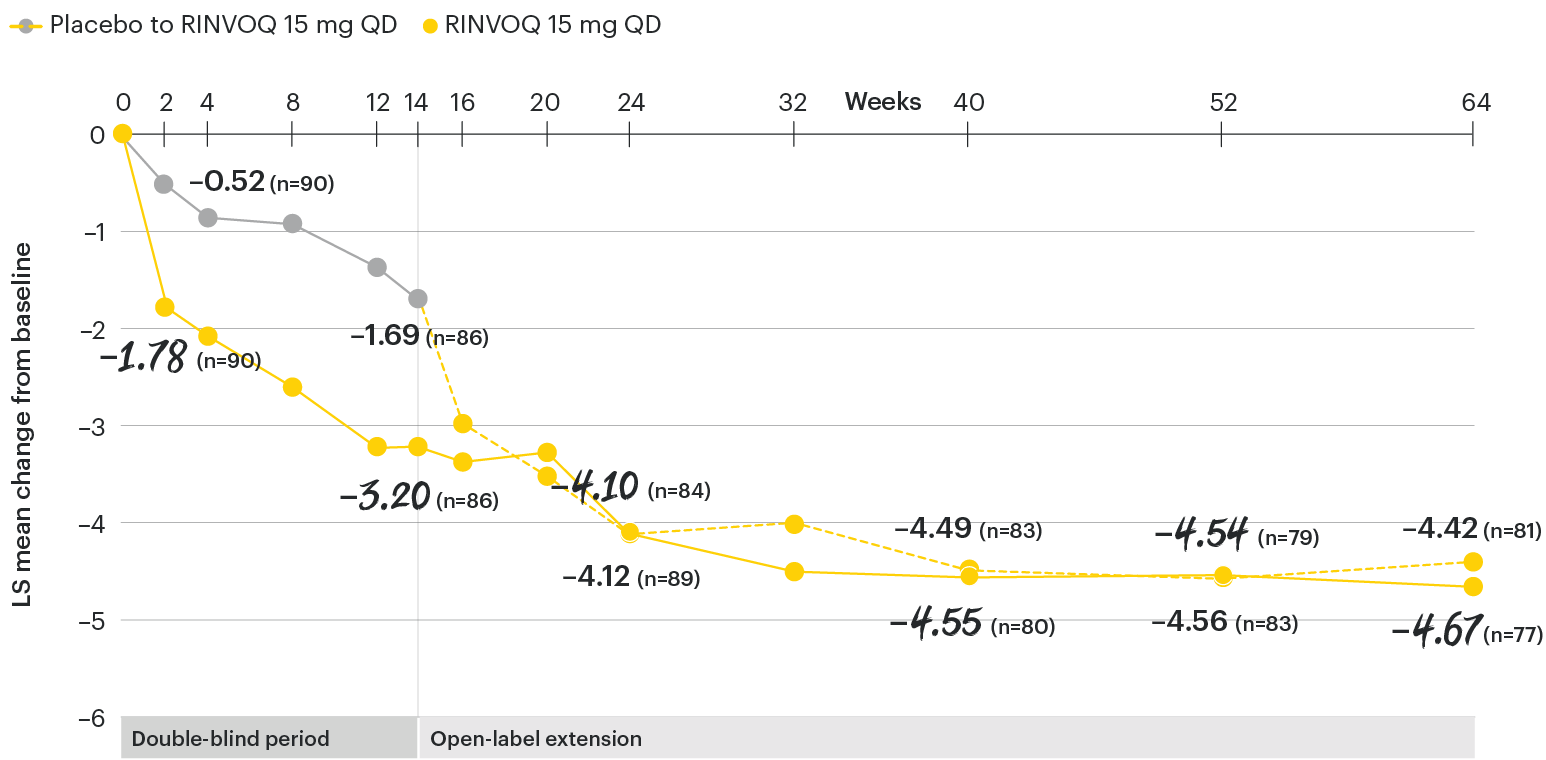
*Back pain defined on a numerical rating scale (0–10) based on the following question: “What is the amount of back pain that you experienced at any time during the last week?”
At baseline, mean back pain scores were 6.7 for placebo and 6.8 for RINVOQ 15 mg QD.2
All patients randomized to placebo received open-label RINVOQ 15 mg QD starting from Week 14. Data shown are from the 64-week analysis of SELECT-AXIS 1 using MMRM for missing data. Because MMRM utilizes all available data to estimate a value for missing data, there may be small differences in the data out to Week 64 when compared with earlier analyses.
DATA LIMITATIONS: Data not labeled as a ranked primary or secondary endpoint were prespecified, however they were not ranked or controlled for multiplicity; therefore, treatment differences could represent chance findings. No conclusions regarding these comparisons can be made.
LS: least squares; MMRM: mixed-effect model for repeated measures; QD: once daily.
In patients with active AS and an inadequate response to conventional therapy
Change in patient global assessment of disease activity† from baseline over time
SELECT-AXIS 1: LS mean change in patient global assessment from baseline to Week 64 (MMRM)3
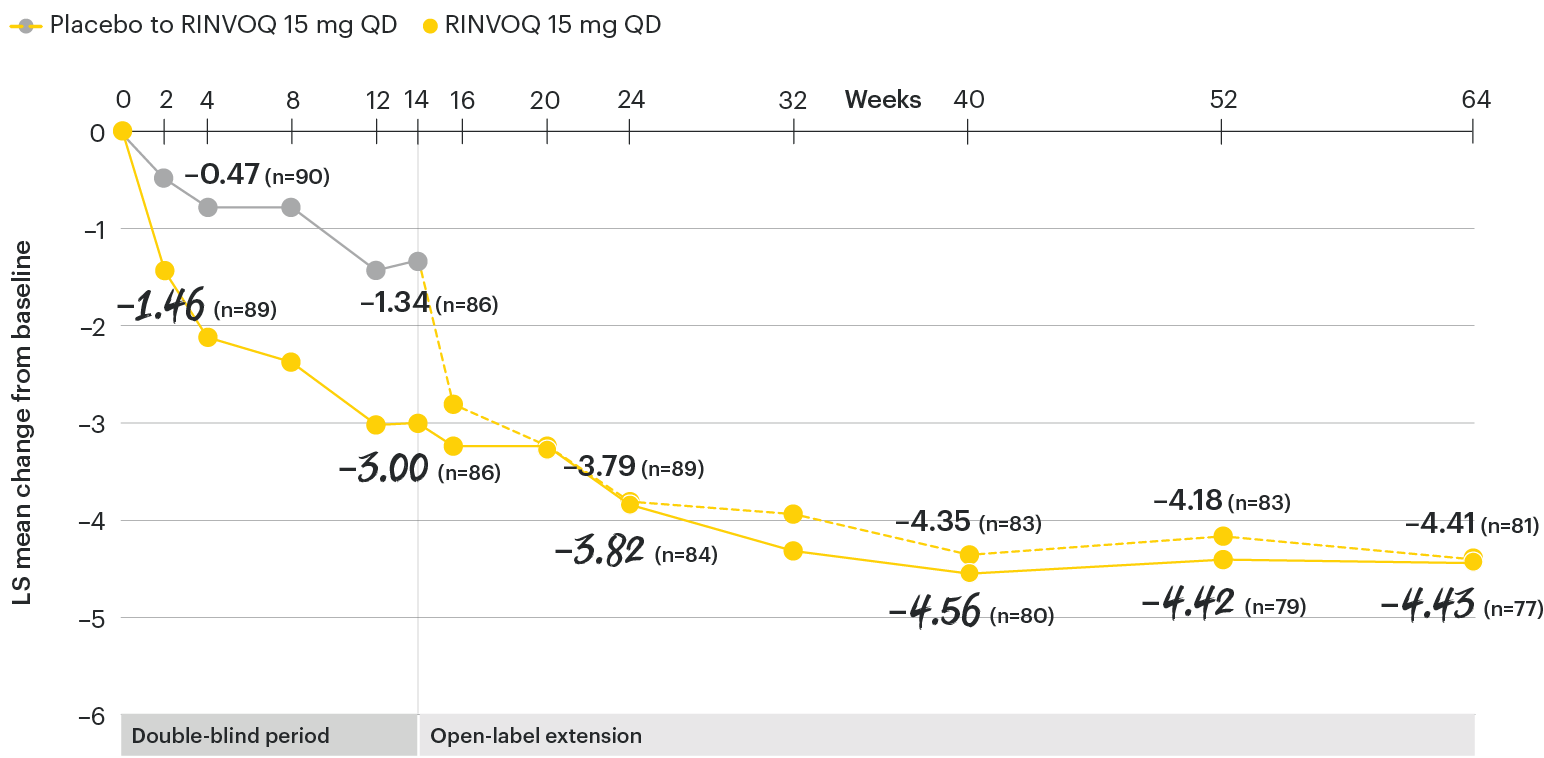
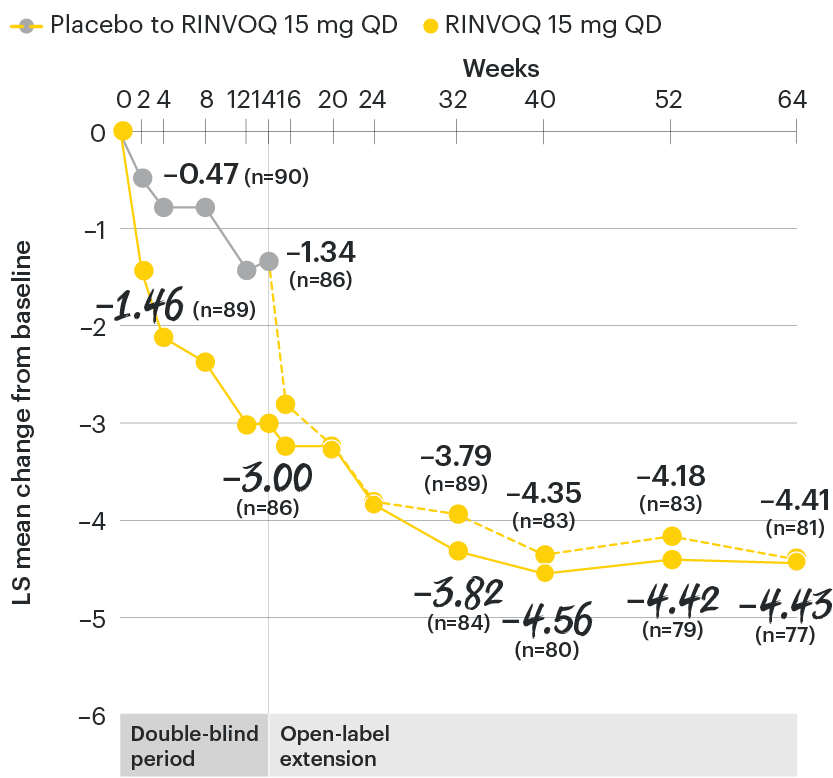
†Patient's global assessment of disease activity was measured on a numerical rating scale ranging from 0 (no activity) to 10 (severe activity).5
At baseline, mean patient global assessment of disease activity scores were 6.8 for placebo and 6.6 for RINVOQ 15 mg QD.2
All patients randomized to placebo received open-label RINVOQ 15 mg QD starting from Week 14. Data shown are from the 64-week analysis of SELECT-AXIS 1 using MMRM for missing data. Because MMRM utilizes all available data to estimate a value for missing data, there may be small differences in the data out to Week 64 when compared with earlier analyses.
DATA LIMITATIONS: Data not labeled as a ranked primary or secondary endpoint were prespecified, however they were not ranked or controlled for multiplicity; therefore, treatment differences could represent chance findings. No conclusions regarding these comparisons can be made.
LS: least squares; MMRM: mixed-effect model for repeated measures; QD: once daily.
In patients with active AS and an inadequate response to conventional therapy
Change in function from baseline over time
SELECT-AXIS 1: LS mean change in BASFI‡ from baseline to Week 64 (MMRM)3
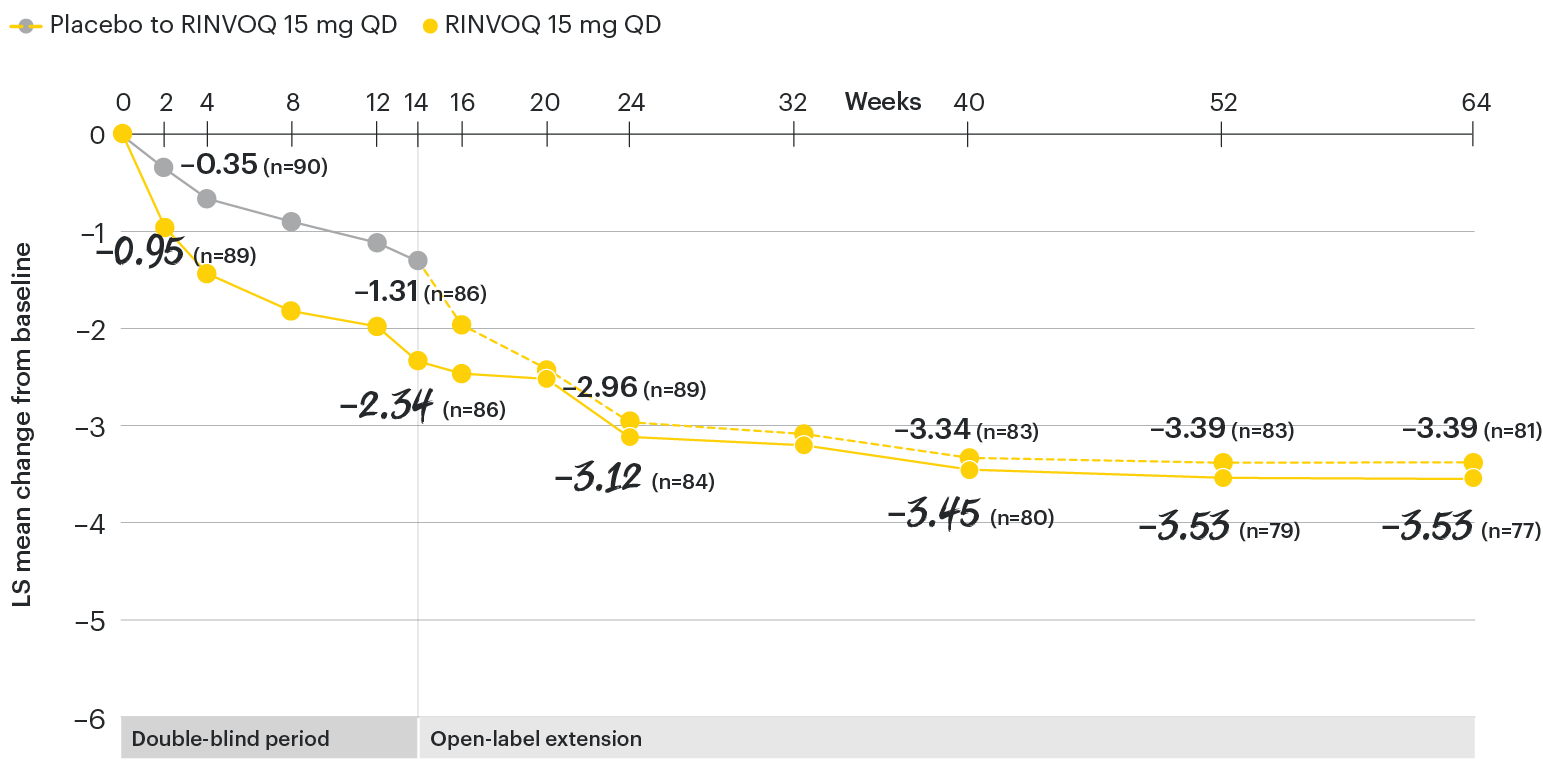
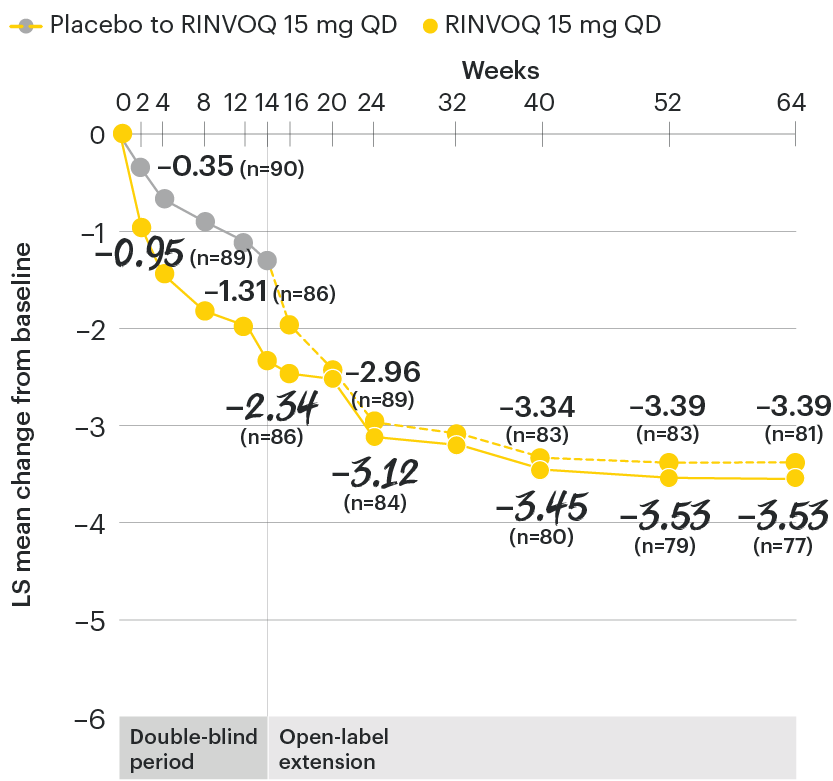
‡Function was measured by the Bath Ankylosing Spondylitis Functional Index (BASFI) which consists of 10 items assessing participants' ability to perform activities on a numerical rating scale ranging from 0 (easy) to 10 (impossible).5
At baseline, mean BASFI scores were 5.5 for placebo and 5.4 for RINVOQ 15 mg QD.2
All patients randomized to placebo received open-label RINVOQ 15 mg QD starting from Week 14. Data shown are from the 64-week analysis of SELECT-AXIS 1 using MMRM for missing data. Because MMRM utilizes all available data to estimate a value for missing data, there may be small differences in the data out to Week 64 when compared with earlier analyses.
DATA LIMITATIONS: Data not labeled as a ranked primary or secondary endpoint were prespecified, however they were not ranked or controlled for multiplicity; therefore, treatment differences could represent chance findings. No conclusions regarding these comparisons can be made.
BASFI: Bath Ankylosing Spondylitis Functional Index; LS: least squares; MMRM: mixed-effect model for repeated measures; QD: once daily.
In patients with active AS and an inadequate response to conventional therapy
Change in inflammation from baseline over time
SELECT-AXIS 1: LS mean change in inflammation§ from baseline to Week 64 (MMRM)3
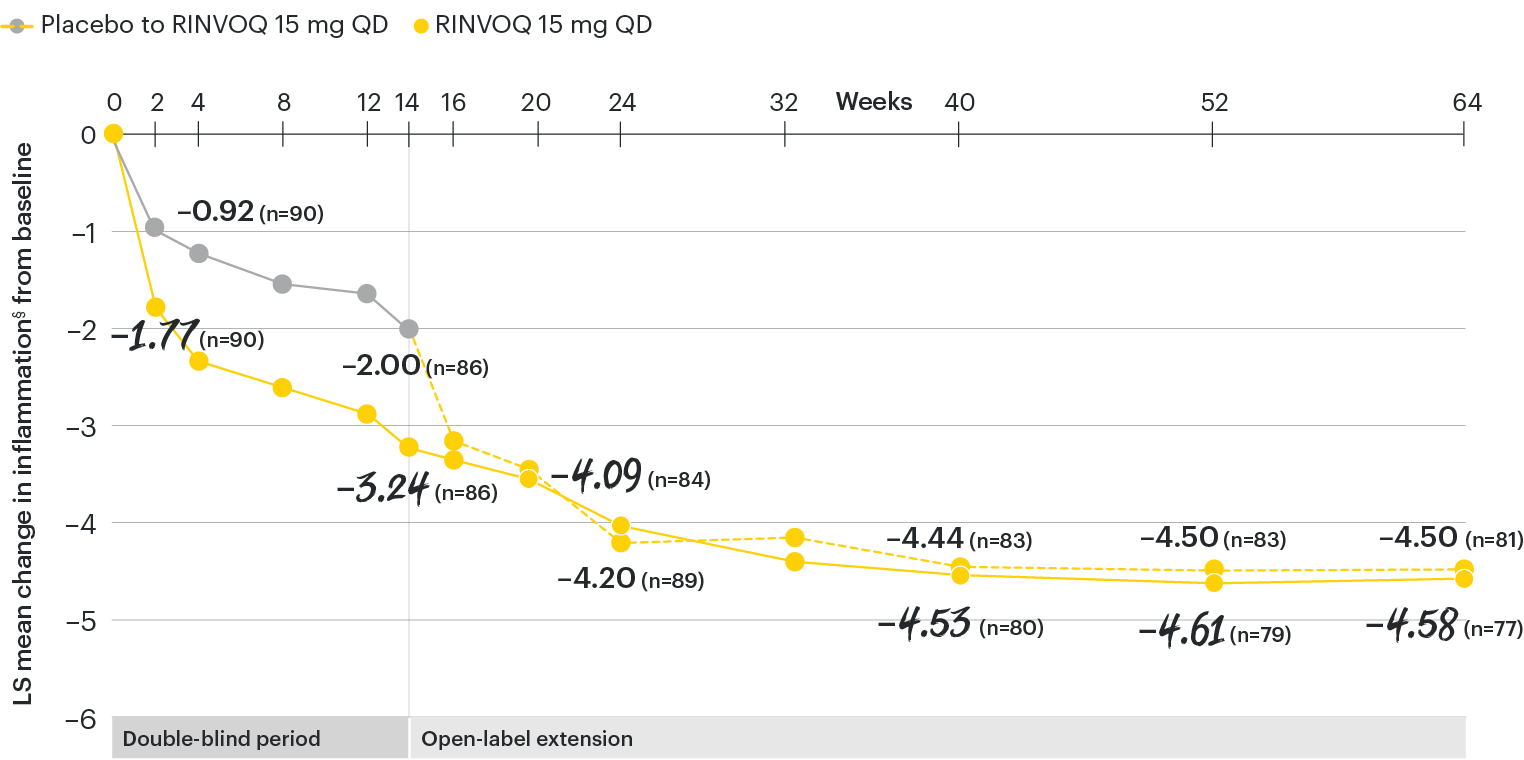
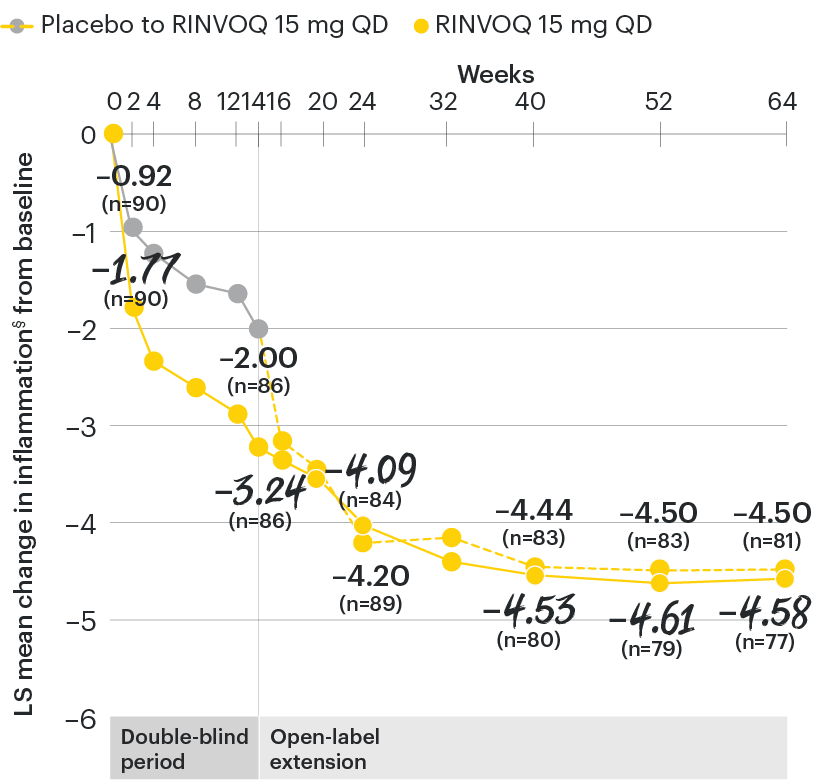
§Inflammation is represented by the mean of the two morning stiffness-related BASDAI NRS scores (mean of items 5 and 6 of the BASDAI [0 to 10]). BASDAI item 5 is based on the following question: “How would you describe the overall level of morning stiffness you have had from the time you wake up?” BASDAI item 6 is based on the following question: “How long does your morning stiffness last from the time you wake up?”
At baseline, mean inflammation§ scores were 6.7 for placebo and 6.5 for RINVOQ 15 mg QD.2
All patients randomized to placebo received open-label RINVOQ 15 mg QD starting from Week 14. Data shown are from the 64-week analysis of SELECT-AXIS 1 using MMRM for missing data. Because MMRM utilizes all available data to estimate a value for missing data, there may be small differences in the data out to Week 64 when compared with earlier analyses.
DATA LIMITATIONS: Data not labeled as a ranked primary or secondary endpoint were prespecified, however they were not ranked or controlled for multiplicity; therefore, treatment differences could represent chance findings. No conclusions regarding these comparisons can be made.
BASDAI: Bath Ankylosing Spondylitis Disease Activity Index; LS: least squares; MMRM: mixed-effect model for repeated measures; NRS: numeric rating scale; QD: once daily.

In patients with active AS and an inadequate response to conventional therapy
Significant improvement in disease activity
SELECT-AXIS 1: BASDAI50 at Week 14 (NRI)1,2
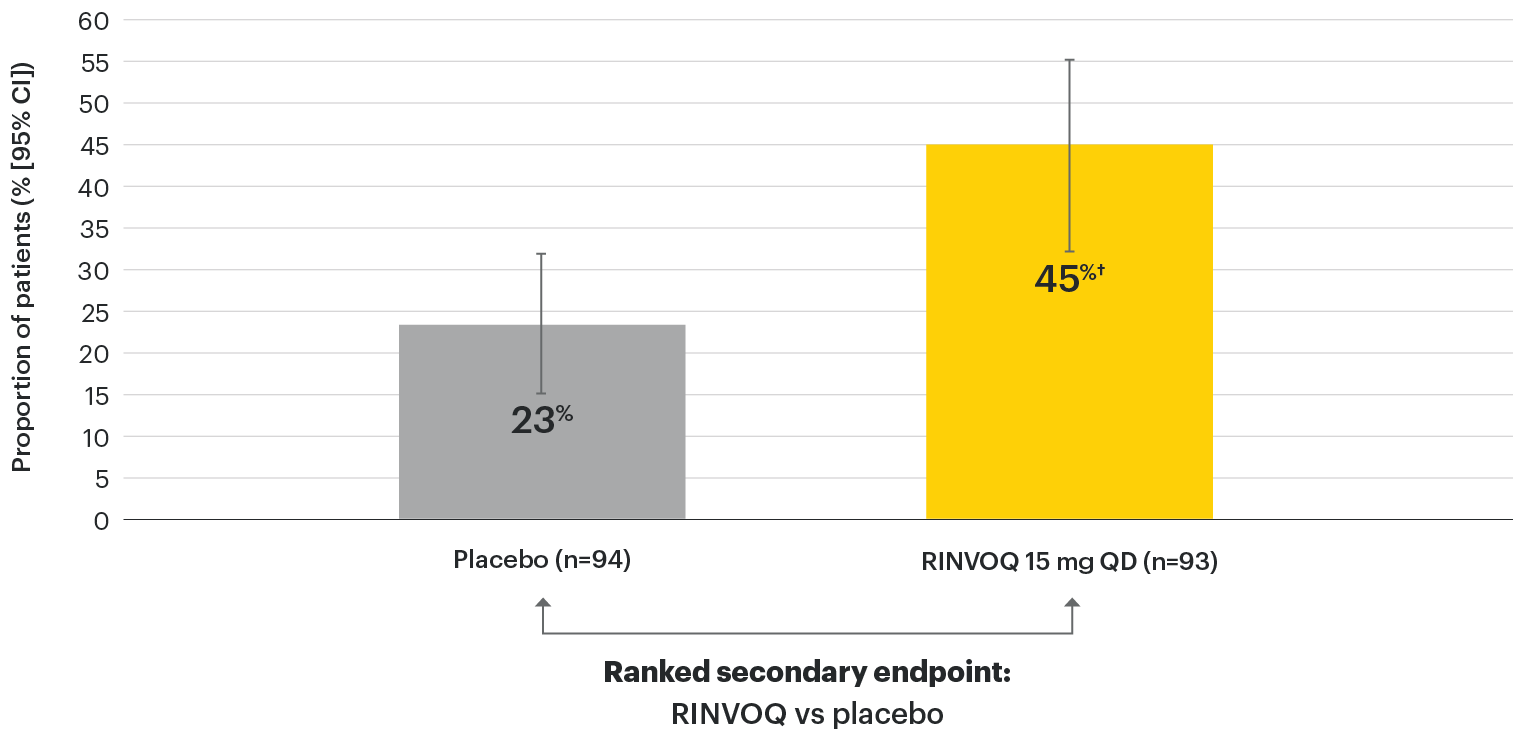
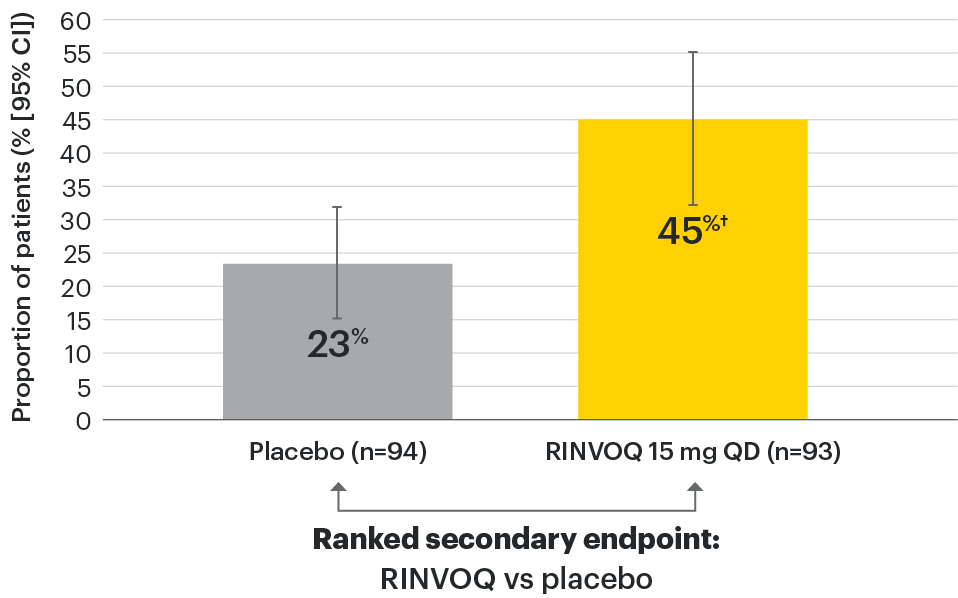
†P≤0.01 vs placebo, statistically significant in multiplicity-controlled analysis.
95% confidence intervals are displayed as error bars in the chart. Missing data were handled using NRI.
BASDAI50: at least 50% improvement in Bath Ankylosing Spondylitis Disease Activity Index; CI: confidence interval; NRI: nonresponder imputation; QD: once daily.
NOTE TO AFFILIATES: Please evaluate references and presentation of data to Week 64, according to local policy, codes and regulations.
In patients with active AS and an inadequate response to conventional therapy
BASDAI50 response through 64 weeks
SELECT-AXIS 1: BASDAI50 response rate to Week 64 (NRI)3


All patients randomized to placebo received open-label RINVOQ 15 mg QD starting from Week 14. Data shown are from the 64-week analysis of SELECT-AXIS 1.
95% confidence intervals are displayed as error bars in the chart, where available. Missing data were handled using NRI.
DATA LIMITATIONS: Data not labeled as a ranked primary or secondary endpoint were prespecified, however they were not ranked or controlled for multiplicity; therefore, treatment differences could represent chance findings. No conclusions regarding these comparisons can be made.
BASDAI50: at least 50% improvement in Bath Ankylosing Spondylitis Disease Activity Index; CI: confidence interval; NRI: nonresponder imputation; QD: once daily.

In patients with active AS and an inadequate response to conventional therapy
Improvement across multiple measures of pain
SELECT-AXIS 1: LS mean change in patient-reported measures of pain at Week 14 (MMRM)3
NOTES TO AFFILIATES: Please evaluate references and presentation of data at Wk 14 and to 64, according to local policy, codes and regulations.
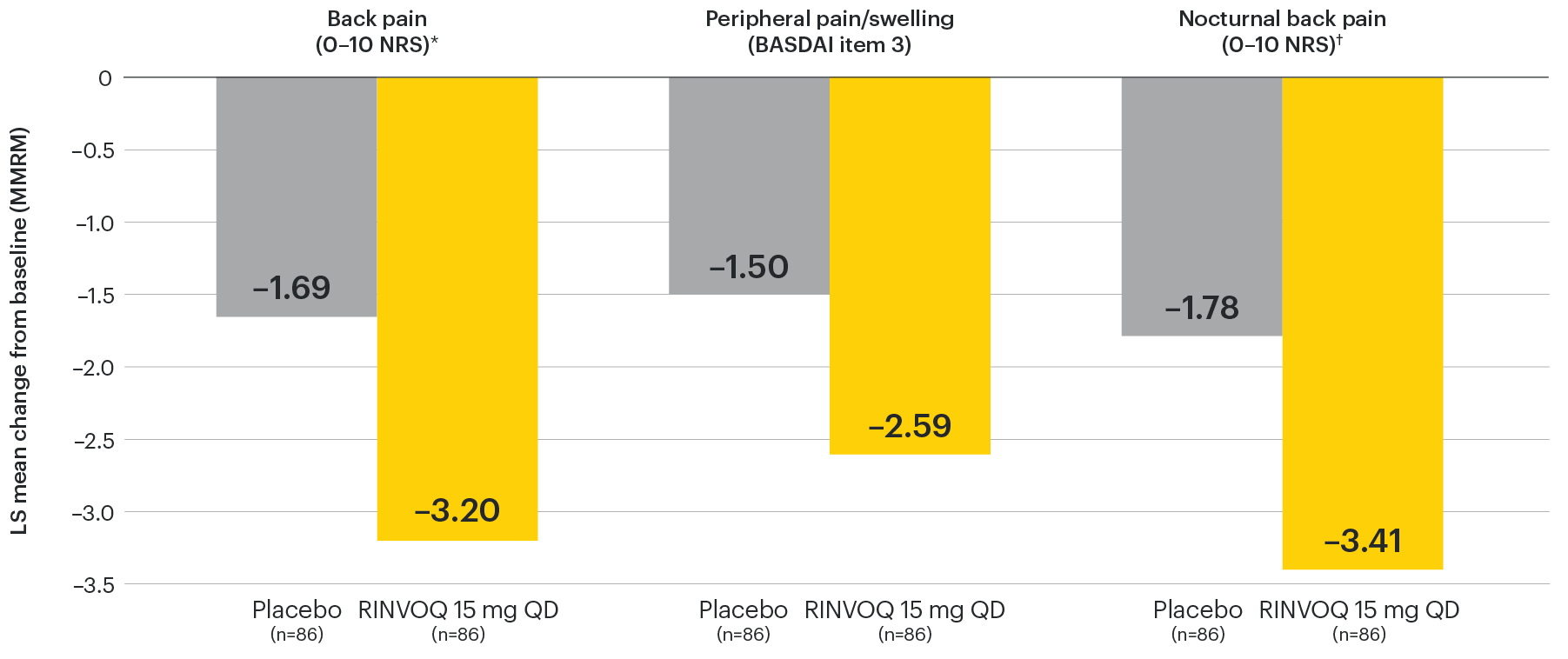
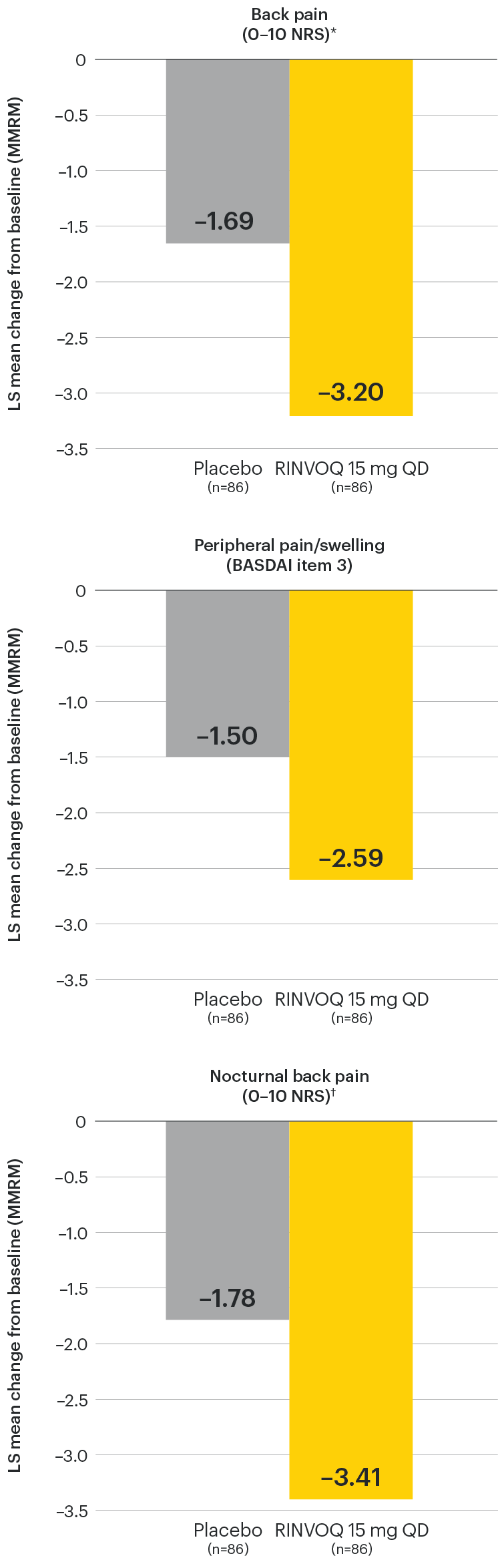
DATA LIMITATIONS: Data not labeled as a ranked primary or secondary endpoint were prespecified, however they were not ranked or controlled for multiplicity; therefore, treatment differences could represent chance findings. No conclusions regarding these comparisons can be made.
*Back pain domain of the ASAS40 measure defined on a numerical rating scale (0–10) based on the following question: “What is the amount of back pain that you experienced at any time during the last week?”
†Nocturnal back pain defined on a numerical rating scale (0–10) based on the following question: “What is the amount of back pain at night that you experienced during the last week?”
Data shown are from the 64-week analysis of SELECT-AXIS 1 using MMRM for missing data. Because MMRM utilizes all available data to estimate a value for missing data, there may be small differences in the data when compared with earlier analyses.
ASAS40: at least 40% improvement in Assessment of Ankylosing Spondylitis International Working Group criteria; BASDAI: Bath Ankylosing Spondylitis Disease Activity Index; MMRM: mixed-effect model for repeated measures; NRS: numeric rating scale; QD: once daily.
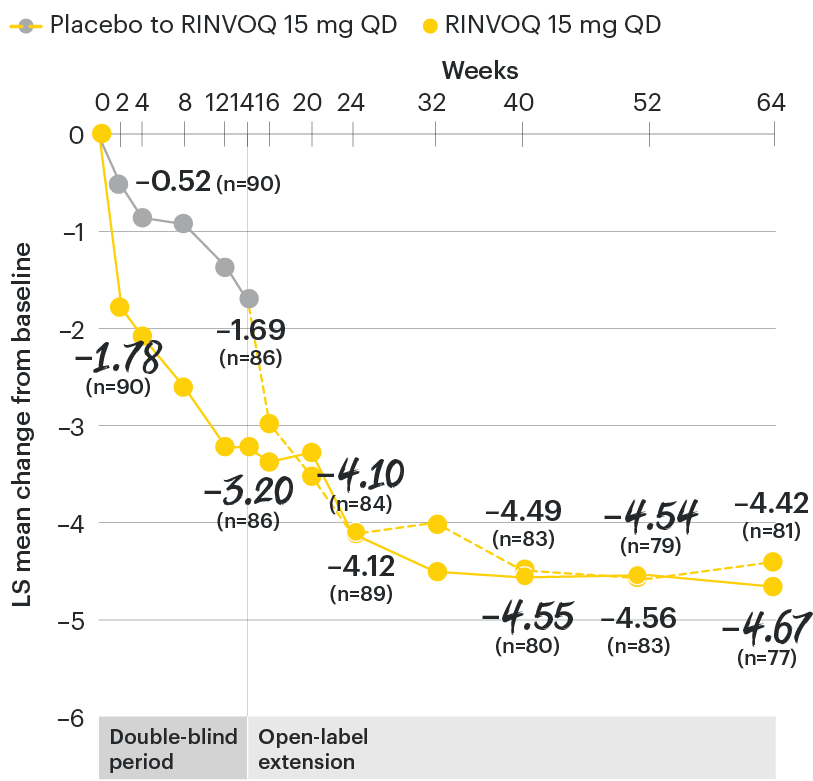
In patients with active AS and an inadequate response to conventional therapy
Change in back pain from baseline over time
SELECT-AXIS 1: LS mean change in patient-reported back pain* from baseline to Week 64 (MMRM)3
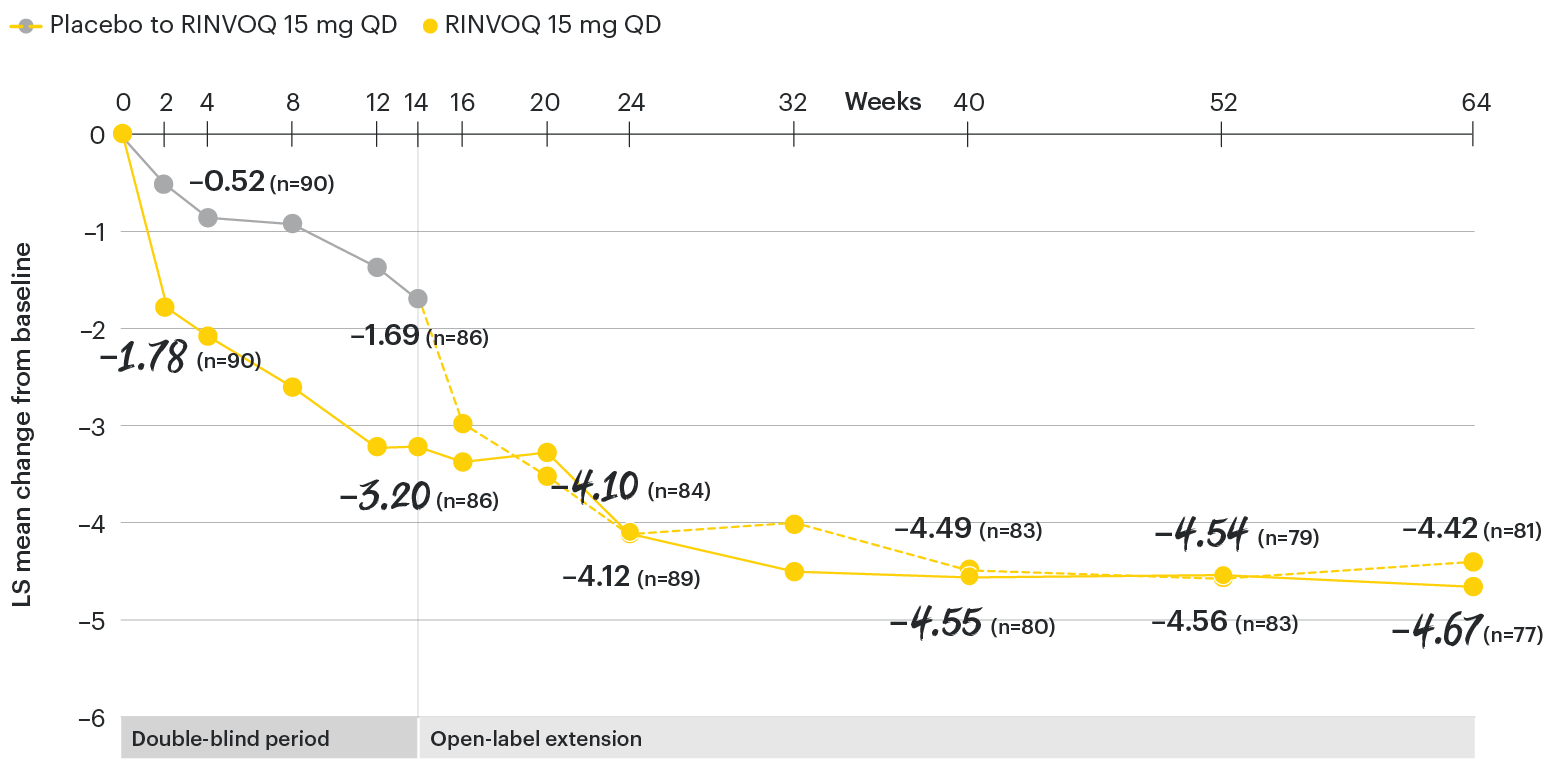
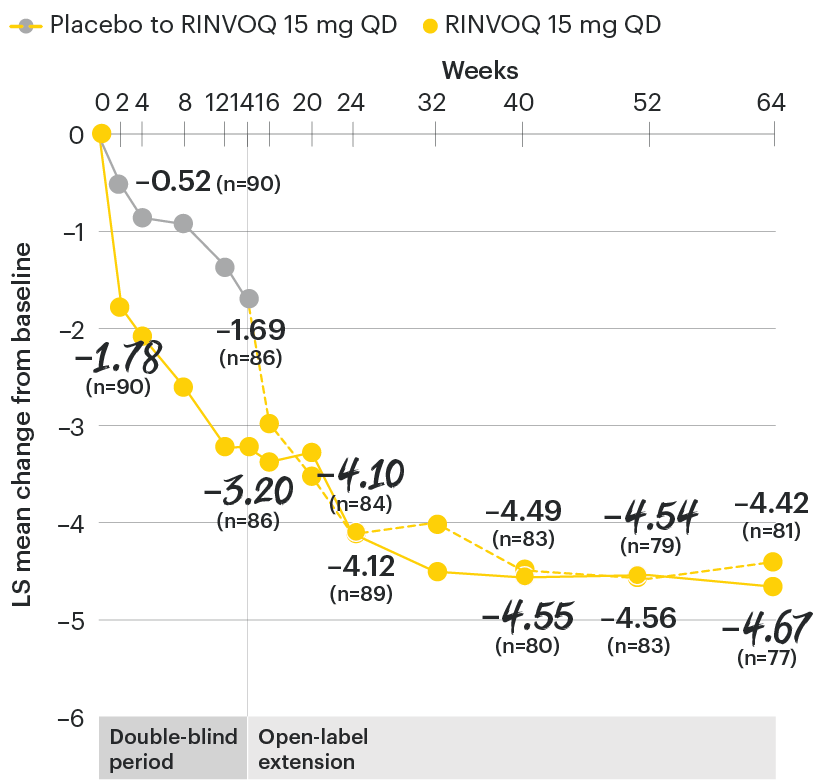
*Back pain defined on a numerical rating scale (0–10) based on the following question: “What is the amount of back pain that you experienced at any time during the last week?”
At baseline, mean back pain scores were 6.7 for placebo and 6.8 for RINVOQ 15 mg QD.
All patients randomized to placebo received open-label RINVOQ 15 mg QD starting from Week 14. Data shown are from the 64-week analysis of SELECT-AXIS 1 using MMRM for missing data. Because MMRM utilizes all available data to estimate a value for missing data, there may be small differences in the data out to Week 64 when compared with earlier analyses.
DATA LIMITATIONS: Data not labeled as a ranked primary or secondary endpoint were prespecified, however they were not ranked or controlled for multiplicity; therefore, treatment differences could represent chance findings. No conclusions regarding these comparisons can be made.
LS: least squares; MMRM: mixed-effect model for repeated measures; QD: once daily.
In patients with active AS and an inadequate response to conventional therapy
Change in peripheral pain from baseline over time
SELECT-AXIS 1: LS mean change in peripheral pain/swelling (BASDAI item 3) from baseline to Week 64 (MMRM)3


All patients randomized to placebo received open-label RINVOQ 15 mg QD starting from Week 14. Data shown are from the 64-week analysis of SELECT-AXIS 1 using MMRM for missing data. Because MMRM utilizes all available data to estimate a value for missing data, there may be small differences in the data out to Week 64 when compared with earlier analyses.
DATA LIMITATIONS: Data not labeled as a ranked primary or secondary endpoint were prespecified, however they were not ranked or controlled for multiplicity; therefore, treatment differences could represent chance findings. No conclusions regarding these comparisons can be made.
BASDAI: Bath Ankylosing Spondylitis Disease Activity Index; LS: least squares; MMRM: mixed-effect model for repeated measures; QD: once daily.
In patients with active AS and an inadequate response to conventional therapy
Change in nocturnal back pain from baseline over time
SELECT-AXIS 1: LS mean change in nocturnal back pain (0–10 NRS)† from baseline to Week 64 (MMRM)3


†Nocturnal back pain defined on a numerical rating scale (0–10) based on the following question: “What is the amount of back pain at night that you experienced during the last week?”
All patients randomized to placebo received open-label RINVOQ 15 mg QD starting from Week 14. Data shown are from the 64-week analysis of SELECT-AXIS 1 using MMRM for missing data. Because MMRM utilizes all available data to estimate a value for missing data, there may be small differences in the data out to Week 64 when compared with earlier analyses.
DATA LIMITATIONS: Data not labeled as a ranked primary or secondary endpoint were prespecified, however they were not ranked or controlled for multiplicity; therefore, treatment differences could represent chance findings. No conclusions regarding these comparisons can be made.
LS: least squares; MMRM: mixed-effect model for repeated measures; QD: once daily.

In patients with active AS and an inadequate response to conventional therapy
Significant improvement in signs of active spinal inflammation
SELECT-AXIS 1: Change in SPARCC-MRI score from baseline to Week 14 (MMRM)1,2
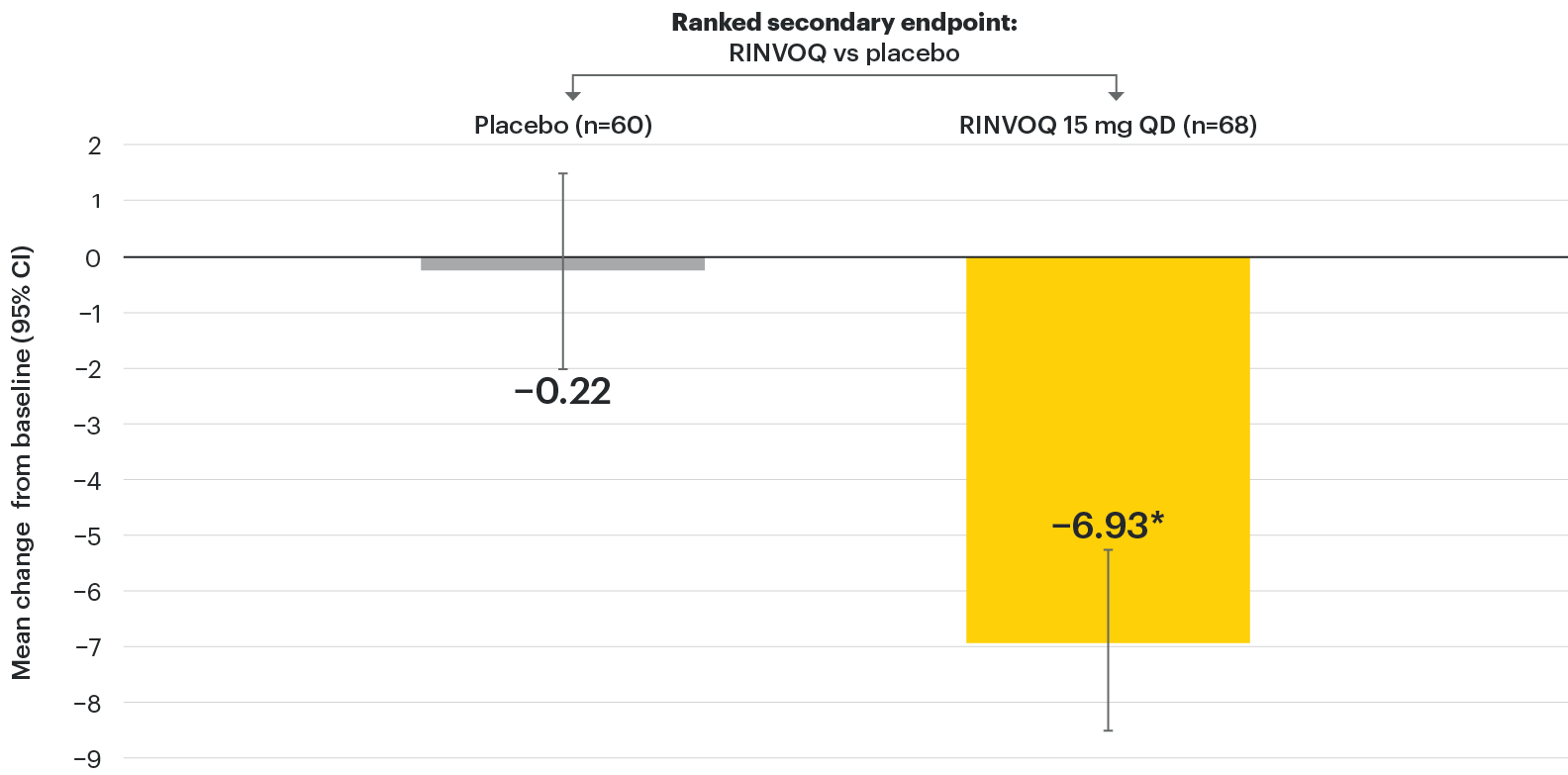
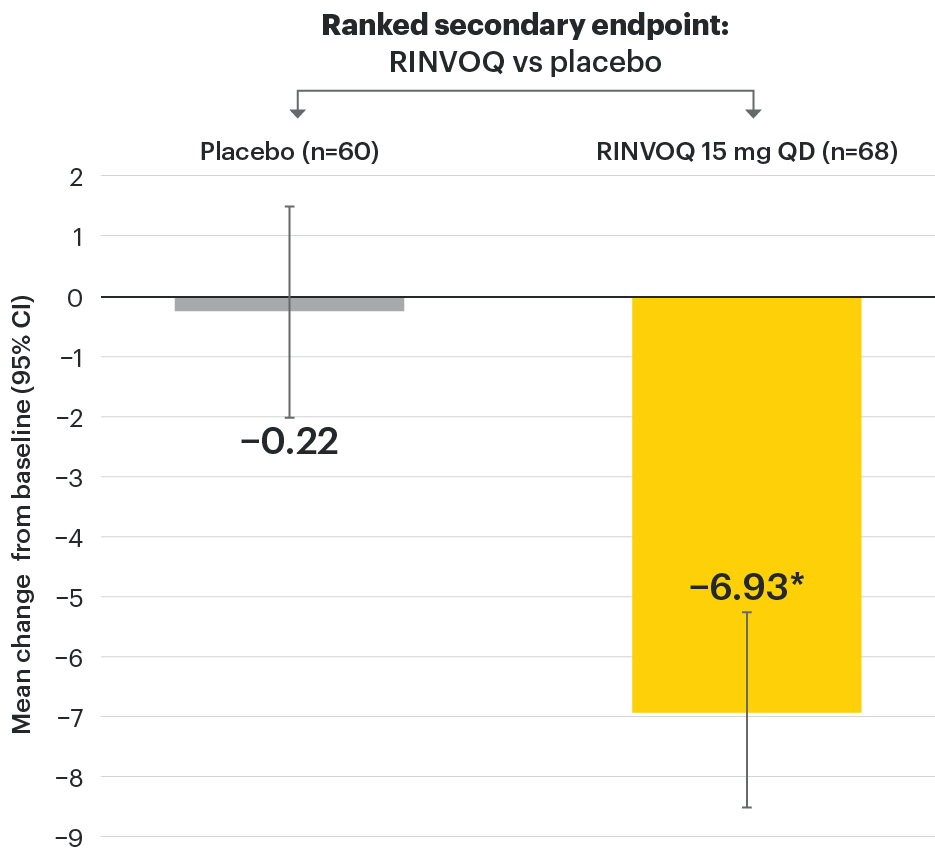
*P≤0.001 vs placebo, statistically significant in multiplicity-controlled analysis.
The SPARCC-MRI assessment population as prespecified in the statistical analysis plan (baseline included MRI data ≤3 days after first dose of study drug and Week 14 included MRI data up to first dose of Period 2 study drug).
95% confidence intervals are displayed as error bars in the chart. Missing data were handled using MMRM.
CI: confidence interval; MMRM: mixed-effect model for repeated measures; QD: once daily; SPARCC MRI: Spondyloarthritis Research Consortium of Canada magnetic resonance imaging.


In patients with active AS and an inadequate response to conventional therapy
The safety profile of RINVOQ in AS was consistent with previously reported results in RA1-3
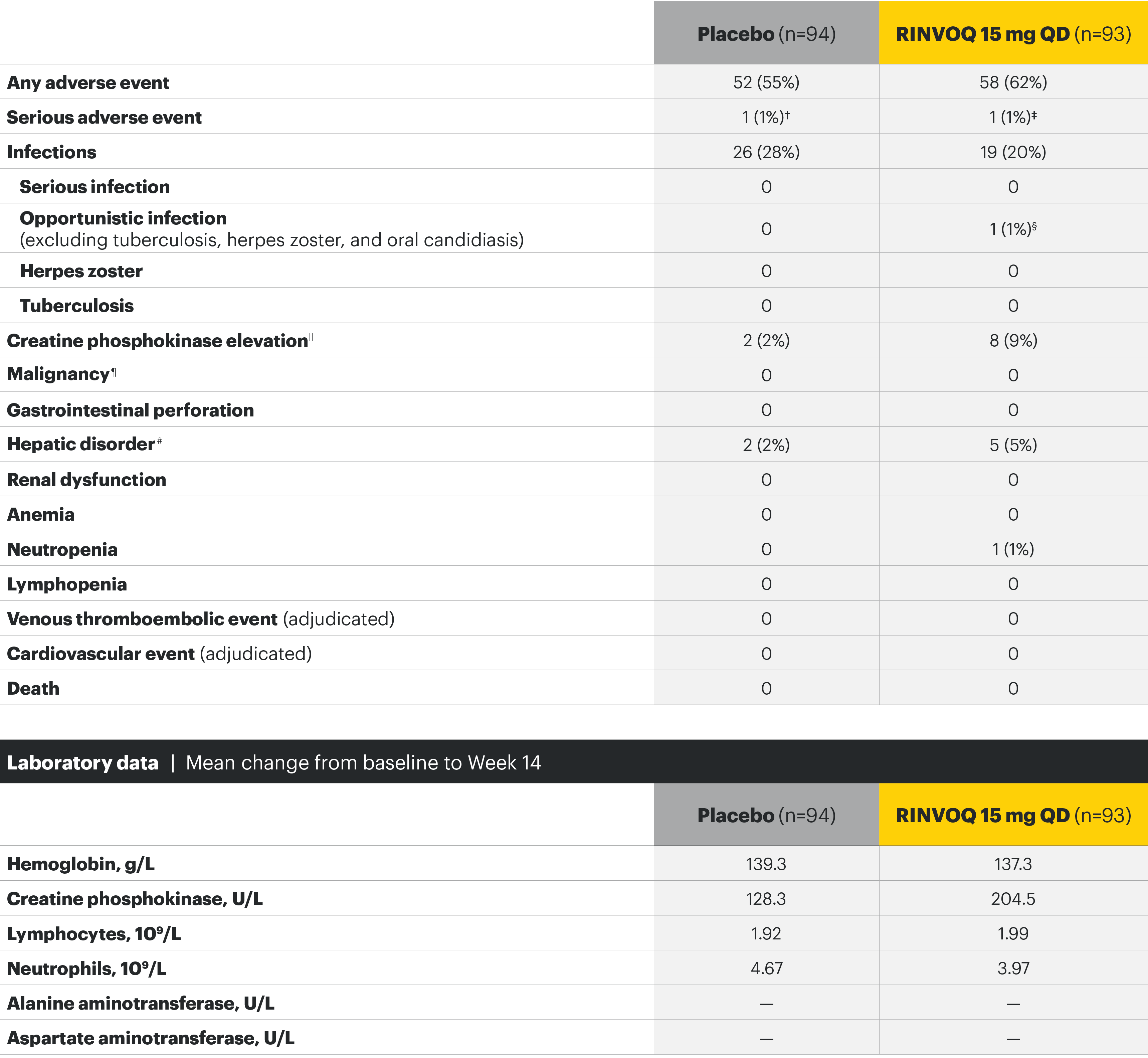
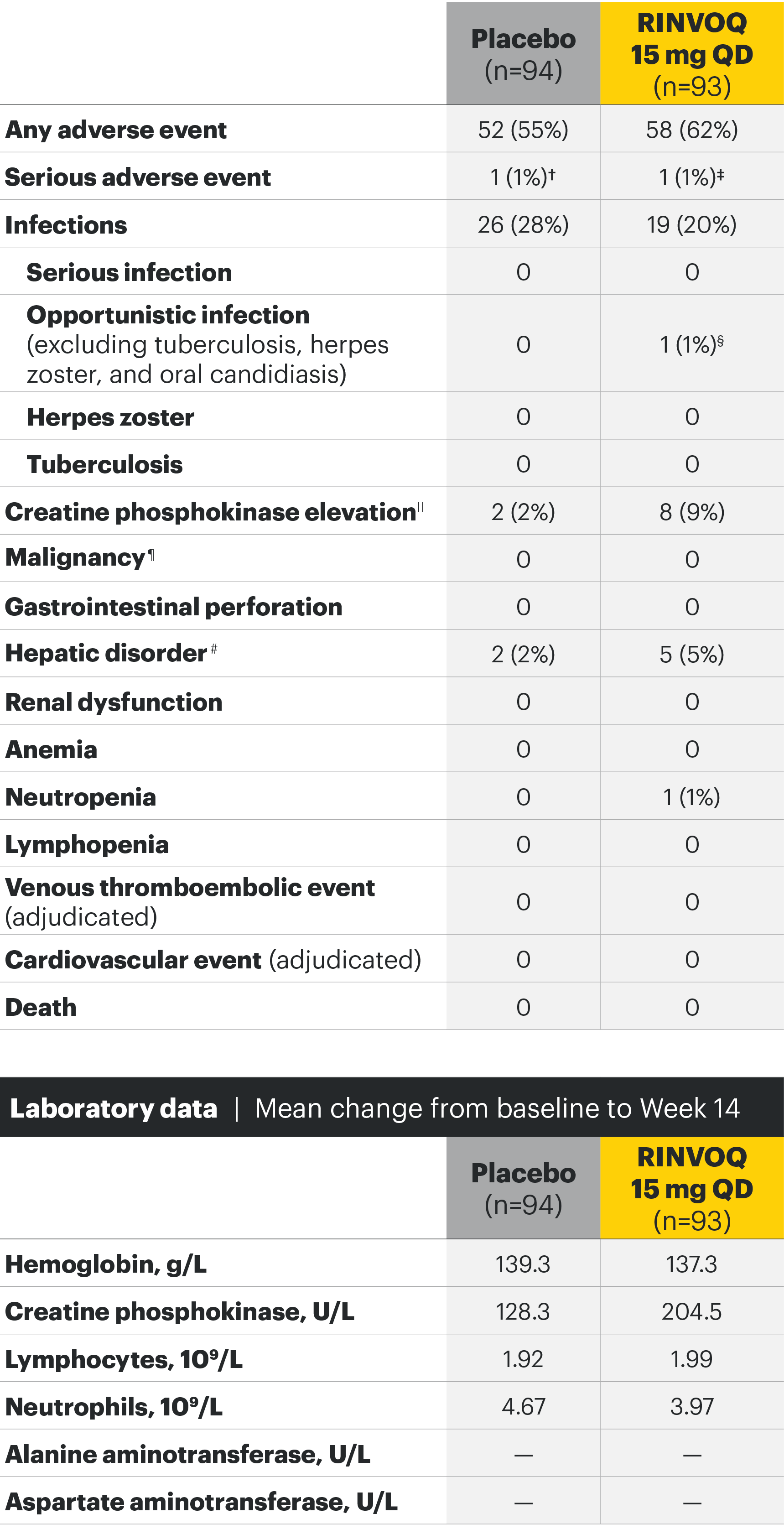
SELECT-AXIS 1: Adverse events through Week 14 and the Week 64 data cut2,3*
Week 14 TEAEs, n (%)2
Week 64 data cut TEAEs (per 100 PYs)3
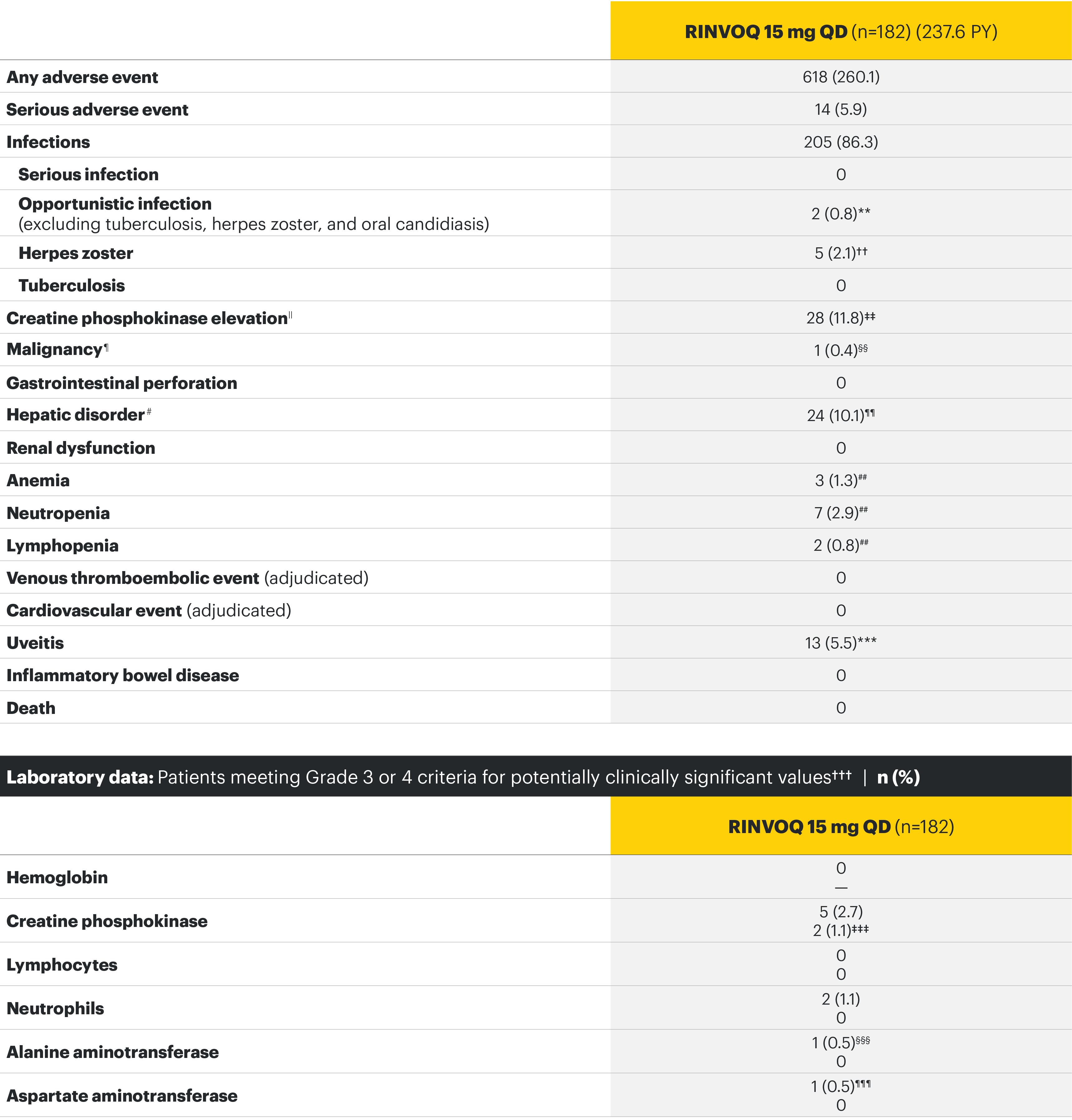
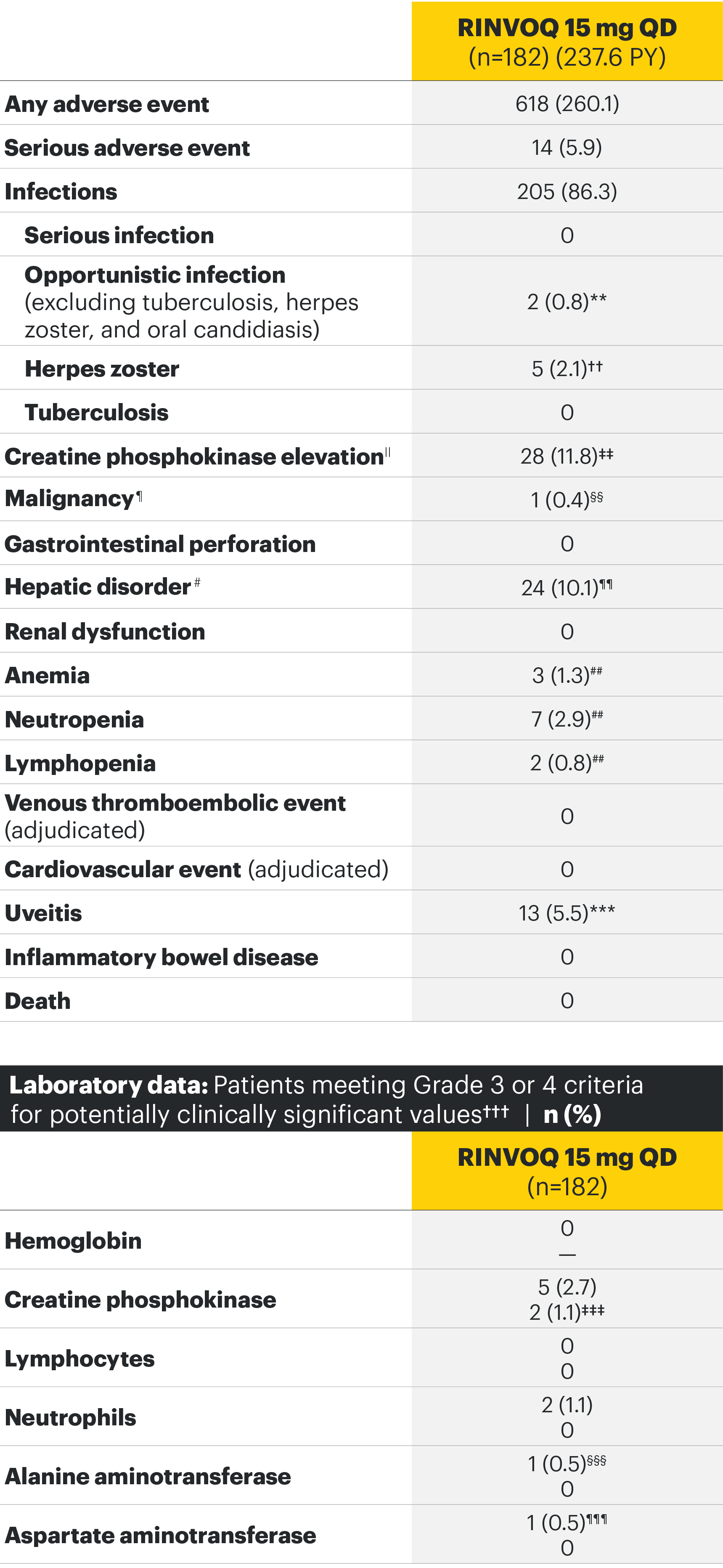
Most common adverse events: In SELECT-AXIS 1, the most common AE at Week 14 was increased creatine phosphokinase.2 At the Week 64 data cut, the most common AEs were nasopharyngitis, increased blood creatine phosphokinase, and upper respiratory tract infection.3
For the safety analysis at the Week 64 data cut, the RINVOQ 15-mg group included patients who were originally randomized to placebo and switched to RINVOQ 15 mg at Week 14.
Overall, the safety profile was consistent with the safety profile observed in patients with rheumatoid arthritis.3
Adverse events leading to discontinuation of study drug occurred in 3% of patients in the placebo group and 2% in the RINVOQ 15-mg-QD group through Week 142 and in 6.3 E/100 PY in the RINVOQ 15-mg-QD group through the Week 64 data cut.3
*Some patients had longer than 64 weeks of exposure at this data cut.
†Cardiovascular disorder, reported as mild circulation dysregulation.
‡Spinal osteoarthritis, reported as moderate worsening of cervical spondylosis 4/5.
§Esophageal candidiasis in patient with gastroesophageal reflux disease; study drug continued after treatment with fluconazole.
||All asymptomatic through Week 14 except for 1 patient in the placebo group.
¶Including nonmelanoma skin cancer, malignancy other than nonmelanoma skin cancer, and lymphoma.
#All 7 hepatic disorders through Week 14 were based on asymptomatic alanine aminotransferase or aspartate aminotransferase increases, and none led to premature discontinuation of study drug.
**Two nonserious events of esophageal candidiasis in the same 60-year-old patient with a history of gastroesophageal reflux disease, each event was moderate in severity and assessed by the investigator as having a reasonable possibility of being related to study drug; study drug was temporarily interrupted for each event but was restarted.
††Five events in 4 patients; all were nonserious and limited to 1 dermatome, and 1 led to discontinuation of the study drug.
‡‡All events were nonserious, and none led to study drug discontinuation; majority were asymptomatic and based on creatine phosphokinase increases of <4xULN.
§§Squamous cell carcinoma of tongue (stage IVa tumor) in 61-year-old male former smoker (approximately 1 pack per day for 40 years); no reasonable possibility to be related to study drug per investigator.
¶¶Majority based on asymptomatic alanine aminotransferase/aspartate aminotransferase elevations; all were nonserious, and none led to study drug discontinuation.
##All events were nonserious, and none led to study drug discontinuation.
***Includes 13 events in 8 patients; all were nonserious and assessed as having no reasonable possibility to be related to study drug; the majority of events occurred in HLA-B27 positive AS patients with a history of uveitis, were mild or moderate in severity, transient, and resolved with local treatment (corticosteroid eye drop); 1 patient discontinued the study.
†††Hemoglobin: Grade 3 (<80 g/L); creatine phosphokinase: Grade 3 (>5.0xULN–10.0xULN), Grade 4 (>10.0xULN); lymphocytes: Grade 3 (0.2–<0.5 x 109/L), Grade 4 (<0.2 x 109/L); neutrophils: Grade 3 (0.5–<1.0 x 109/L), Grade 4 (<0.5 x 109/L); alanine aminotransferase and aspartate aminotransferase: Grade 3 (>5.0–20.0xULN), Grade 4 (>20.0xULN).
‡‡‡Occurred in young male patients; none led to study drug discontinuation, none met toxicity criteria threshold (none confirmed ≥4 x ULN) and 5 were transient and normalized, including the two grade 4 increases.
§§§ALT normalized after interruption of study drug; study drug was continued.
¶¶¶History of an AST increase and slightly elevated AST at baseline; study drug was not interrupted, and no adverse event was reported with the grade 3 event.

RINVOQ Important Safety Information1
Contraindications
RINVOQ is contraindicated in patients hypersensitive to the active substance or to any of the excipients, in patients with active tuberculosis (TB) or active serious infections, in patients with severe hepatic impairment, and during pregnancy.
Special warnings and precautions for use
Immunosuppressive medicinal products
Use in combination with other potent immunosuppressants is not recommended.
Serious infections
Serious and sometimes fatal infections have been reported in patients receiving upadacitinib. The most frequent serious infections reported included pneumonia and cellulitis. Cases of bacterial meningitis have been reported. Among opportunistic infections, TB, multidermatomal herpes zoster, oral/esophageal candidiasis, and cryptococcosis have been reported with upadacitinib. As there is a higher incidence of infections in patients ≥65 years of age, caution should be used when treating this population.
Viral reactivation
Viral reactivation, including cases of herpes zoster, was reported in clinical studies. The risk of herpes zoster appears to be higher in Japanese patients treated with upadacitinib.
Vaccinations
The use of live, attenuated vaccines during or immediately prior to therapy is not recommended. It is recommended that patients be brought up to date with all immunizations, including prophylactic zoster vaccinations, prior to initiating upadacitinib, in agreement with current immunization guidelines.
Malignancy
The risk of malignancies, including lymphoma is increased in patients with rheumatoid arthritis (RA). Malignancies, including nonmelanoma skin cancer (NMSC), have been reported in patients treated with upadacitinib. Consider the risks and benefits of upadacitinib treatment prior to initiating therapy in patients with a known malignancy other than a successfully treated NMSC or when considering continuing upadacitinib therapy in patients who develop a malignancy.
Hematological abnormalities
Treatment should not be initiated, or should be temporarily interrupted, in patients with hematological abnormalities observed during routine patient management.
Diverticulitis
Upadacitinib should be used with caution in patients with diverticular disease and especially in patients chronically treated with concomitant medications associated with an increased risk of diverticulitis.
Cardiovascular risk
RA patients have an increased risk for cardiovascular disorders. Patients treated with upadacitinib should have risk factors (e.g., hypertension, hyperlipidemia) managed as part of usual standard of care.
Lipids
Upadacitinib treatment was associated with dose-dependent increases in lipid parameters, including total cholesterol, low-density lipoprotein cholesterol, and high-density lipoprotein cholesterol.
Hepatic transaminase elevations
Treatment with upadacitinib was associated with an increased incidence of liver enzyme elevation compared to placebo.
Venous thromboembolisms
Events of deep vein thrombosis (DVT) and pulmonary embolism (PE) have been reported in patients receiving JAK inhibitors, including upadacitinib. Upadacitinib should be used with caution in patients at high risk for DVT/PE.
Adverse reactions
The most commonly reported adverse reactions in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis clinical trials (≥2% of patients in at least one of the indications) with upadacitinib 15 mg were upper respiratory tract infections, blood creatine phosphokinase (CPK) increased, alanine transaminase increased (ALT), bronchitis, nausea, cough, aspartate transaminase increased (AST), and hypercholesterolemia. The most common serious adverse reactions were serious infections.
The safety profile of upadacitinib with long term treatment was generally similar to the safety profile during the placebo-controlled period across indications.
Overall, the safety profile observed in patients with psoriatic arthritis or active ankylosing spondylitis treated with upadacitinib 15 mg was consistent with the safety profile observed in patients with RA.
This is not a complete summary of all safety information.
Please see the RINVOQ Summary of Product Characteristics for complete prescribing information.
You may also be interested in





I want to receive more information
via a product specialist.

I have a question for
Medical Information.

- RINVOQ [Summary of Product Characteristics]. AbbVie Deutschland GmbH & Co. KG.
- van der Heijde D, Song IH, Pangan AL, et al. Efficacy and safety of upadacitinib in patients with active ankylosing spondylitis (SELECT-AXIS 1): a multicentre, randomised, double-blind, placebo-controlled, phase 2/3 trial. Lancet. 2019;394(10214):2108-2117. doi:10.1016/S0140-6736(19)32534-6
- Deodhar A, van der Heijde D, Sieper J, et al. Upadacitinib in active ankylosing spondylitis: 1-year results from the double-blind, placebo-controlled SELECT-AXIS 1 study and open-label extension [published online July 1, 2021]. Arthritis Rheumatol. doi:10.1002/art.41911
- Assessment of SpondyloArthritis international Society (ASAS). ASDAS calculator. https://www.asas-group.org/clinical-instruments/asdas-calculator. Accessed July 12, 2021.
- A study evaluating the safety and efficacy of upadacitinib in adults with active ankylosing spondylitis (SELECT-AXIS 1). ClinicalTrials.gov identifier: NCT03178487. https://clinicaltrials.gov/ct2/show/NCT03178487. Accessed July 13, 2021.