RINVOQ (upadacitinib) is indicated for the treatment of adults with moderately to severely active rheumatoid arthritis (RA) who have had an inadequate response or intolerance to methotrexate (MTX). RINVOQ may be used as monotherapy or in combination with MTX or other nonbiologic disease-modifying antirheumatic drugs (DMARDs).
INTEGRATED SAFETY ANALYSIS

[Affiliates to insert drivers to local asset]
Integrated safety analysis results
Hear Professor Kevin Winthrop discuss results from an integrated safety analysis from the SELECT Phase 3 clinical program in patients with moderate to severe RA.1

[Affiliates to insert drivers to local asset]
Integrated safety analysis study explainer
Watch an overview of the study design and results.
RINVOQ safety profile: Well-characterized in 5 Phase 3 studies
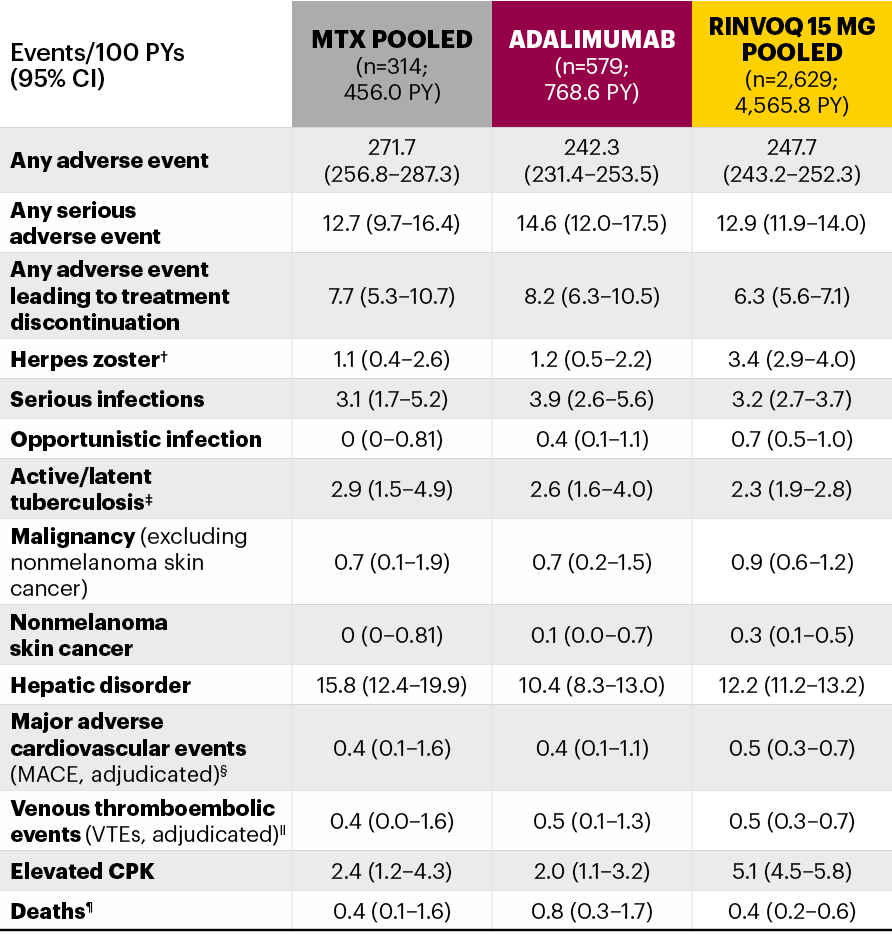
Integrated safety analysis across multiple patient populations up to 53 weeks of exposure1
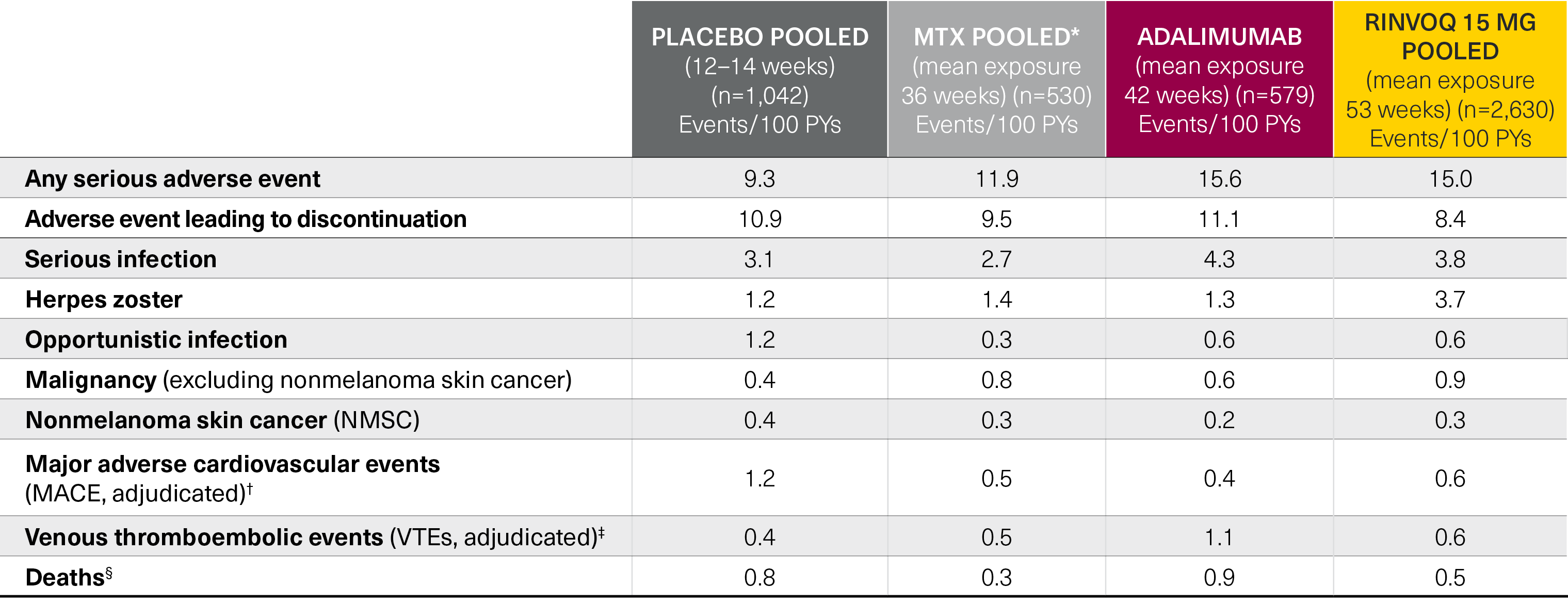
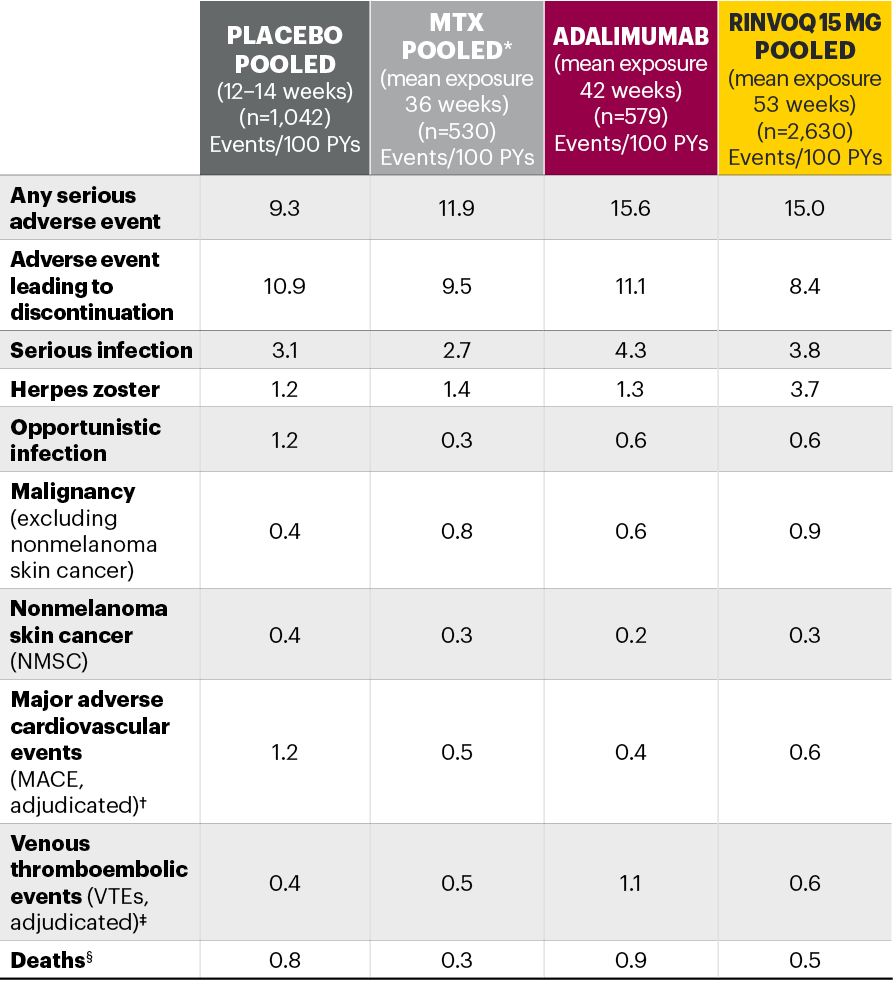
Five pivotal randomized, double-blind, controlled Phase 3 trials of RINVOQ were included in the integrated safety analyses. Placebo data were pooled from 3 placebo-controlled studies with exposures of up to 14 weeks (SELECT-NEXT and SELECT-BEYOND: up to 12 weeks; SELECT-COMPARE: up to 14 weeks). MTX data were pooled from 2 MTX-controlled studies (SELECT-EARLY and SELECT-MONOTHERAPY) with mean exposure of 36 weeks, including a maximum of 14 weeks of MTX treatment in SELECT-MONOTHERAPY. Originator adalimumab data were derived from 1 adalimumab-controlled study (SELECT-COMPARE) with mean exposure of 42 weeks. The RINVOQ 15 mg data were pooled from the above 5 studies with a mean exposure of 53 weeks. A total of 589 (22%) patients received RINVOQ for ≥72 weeks. Patients who switched from placebo, adalimumab, or MTX to RINVOQ were included in the RINVOQ analysis set from the start of RINVOQ, while those who switched from RINVOQ to adalimumab (SELECT-COMPARE) were included in the adalimumab data set from the start of adalimumab. All deaths and cardiovascular events across the clinical program were adjudicated by a blinded external cardiac adjudication committee. The most commonly reported adverse events in the RINVOQ 15 mg pooled group were upper respiratory tract infections, nasopharyngitis, and urinary tract infections.
*MTX pooled includes patients on MTX monotherapy censored at time of switch/addition of RINVOQ.
†MACE was defined as cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke.
‡VTE was defined as deep vein thrombosis and pulmonary embolism.
§Deaths included non–treatment-emergent deaths (3 on RINVOQ and 1 on adalimumab).
MTX: methotrexate; PYs: patient years.
Well-characterized safety profile across multiple patient populations in 5 Phase 3 studies
Integrated safety analysis of RINVOQ up to 3.5 years of exposure2*
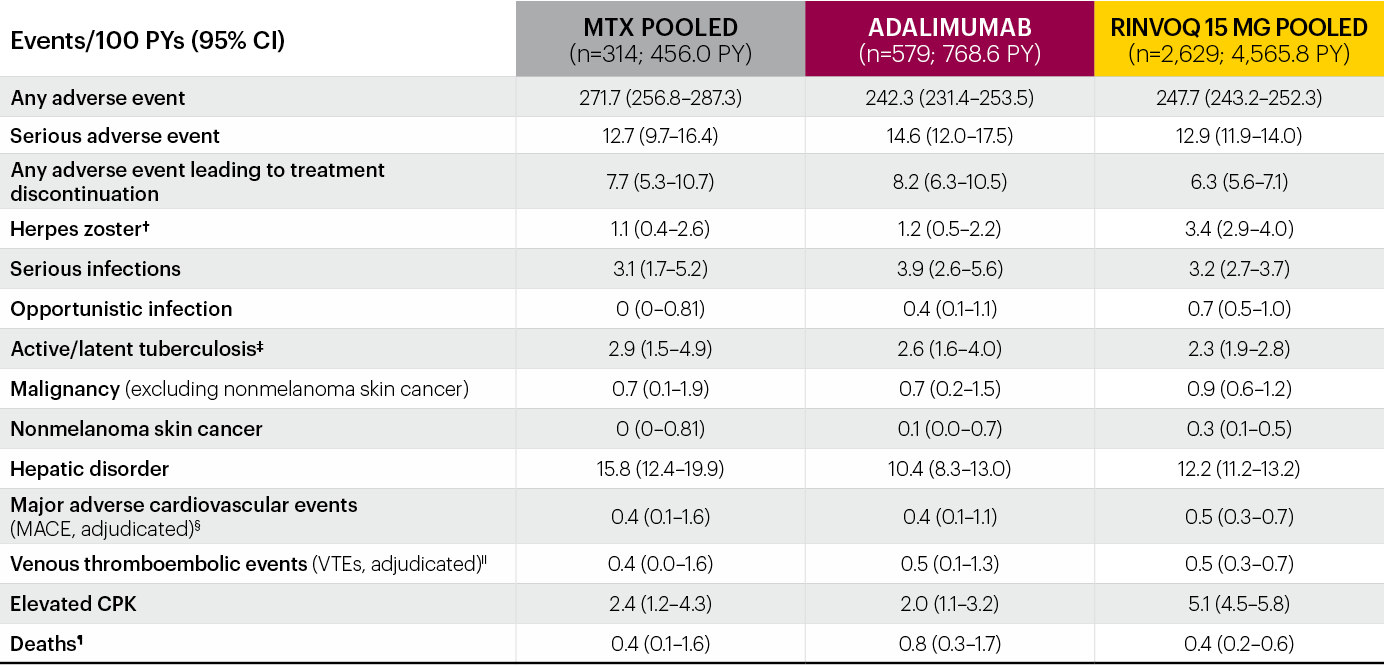
*Patients who switched from placebo, adalimumab, or MTX to UPA were included in the UPA analysis set from the start of UPA, while those who switched from UPA to adalimumab were included in the adalimumab analysis set from the start of adalimumab.
†Most cases of herpes zoster were nonserious and single dermatome.
‡There were 6 cases of active tuberculosis in UPA patients (0.1 E/100 PY) and 1 in an adalimumab patient.
§MACE defined as cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke.
IIVTE defined as deep vein thrombosis and pulmonary embolism.
¶Deaths included non–treatment-emergent deaths.
The most frequent treatment-emergent adverse events (≥5 E/100 PYs) in RINVOQ-treated patients were upper respiratory tract infection, nasopharyngitis, urinary tract infection, bronchitis, increased creatine phosphokinase, increased alanine aminotransferase, worsening RA, and herpes zoster.
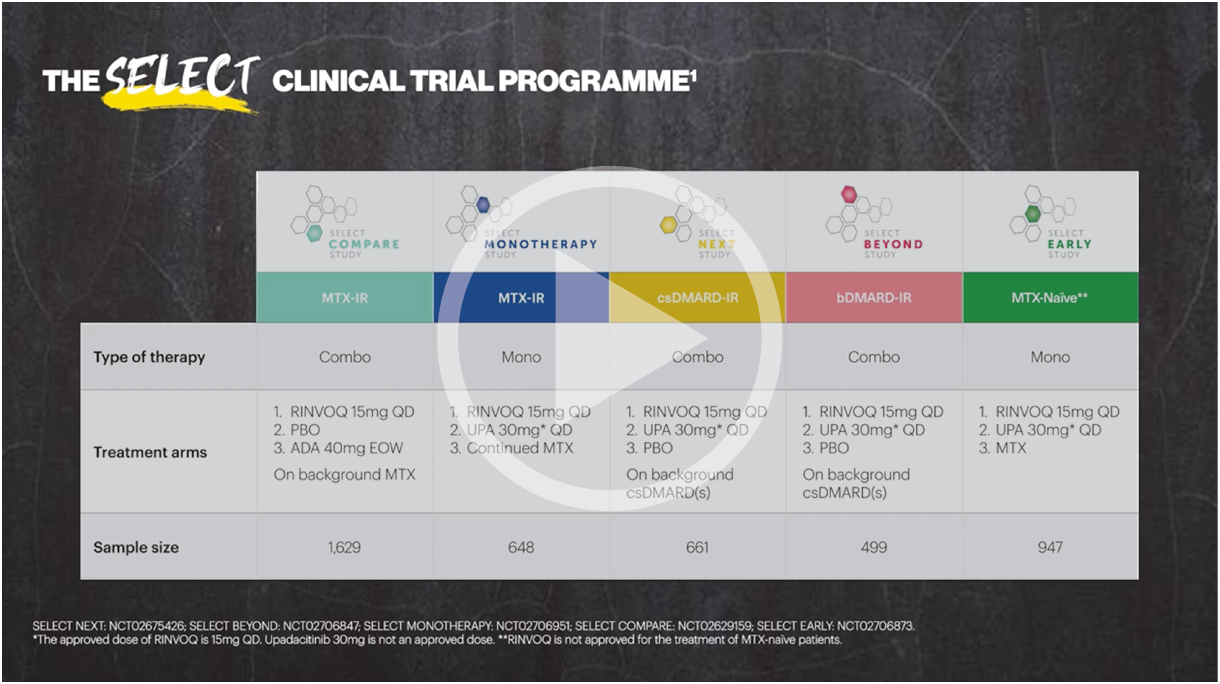
RINVOQ was evaluated from a comprehensive clinical program of 5 pivotal Phase 3 randomized-controlled trials across a spectrum of RA patient populations (SELECT-COMPARE, SELECT-MONOTHERAPY, SELECT-EARLY, SELECT-NEXT, and SELECT-BEYOND). All 5 trials evaluated RINVOQ 15 mg QD and all but SELECT-COMPARE evaluated UPA 30 mg QD. MTX-naive patients receiving MTX monotherapy in SELECT-EARLY and MTX-IR patients receiving adalimumab + MTX in SELECT-COMPARE were included. Two studies evaluated UPA as monotherapy (SELECT-EARLY and SELECT-MONOTHERAPY) while the remaining 3 studies (SELECT-NEXT, SELECT-BEYOND, and SELECT-COMPARE) evaluated UPA in combination with csDMARDs. Patients who switched from placebo, adalimumab, or MTX to UPA were included in the UPA analysis set from the start of UPA treatment, while those who switched from UPA to adalimumab (SELECT-COMPARE) were included in the adalimumab data set from the start of adalimumab treatment. In SELECT-COMPARE, switching from adalimumab to UPA (or vice versa) could occur at Week 14, 18, 22, or 26 based on prespecified criteria. MTX monotherapy was censored at time of rescue to combination therapy (addition of UPA). TEAEs and AESIs were summarized for each treatment group in terms of exposure-adjusted event rates, calculated as the total number of events adjusted for total exposure to UPA and expressed as E/100 PYs. TEAEs were defined as events with an onset date on or after the first dose of study drug and no more than 30 days (70 days for adalimumab) after last dose of study drug in cases of premature discontinuation. Median exposures were 101.9 weeks (maximum 179.7 weeks) in the RINVOQ 15 mg group, 92.6 weeks (maximum 169.0 weeks) in the MTX monotherapy group, and 78.6 weeks (maximum 179.0 weeks) in the adalimumab + MTX group.
AESI: adverse event of special interest; CI: confidence interval; CPK: creatine phosphokinase; E/100 PYs: events per 100 patient-years; MACE: major adverse cardiovascular event; MTX: methotrexate; NMSC: nonmelanoma skin cancer; PY: patient-year; QD: once daily; TEAE: treatment-emergent adverse event; UPA: upadacitinib; VTE: venous thromboembolic event.

Contraindications
- Hypersensitivity to the active substance or to any of the excipients
- Active tuberculosis (TB) or active serious infections
- Severe hepatic impairment
- Pregnancy
Summary of the RINVOQ safety profile
The most commonly reported adverse drug reactions (ADRs) were upper respiratory tract infections, bronchitis, nausea, blood creatine phosphokinase (CPK) increased, and cough. The most common serious adverse reactions were serious infections.3
Adverse drug reactions3
The following table of adverse reactions is based on experience from registrational clinical studies.
Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
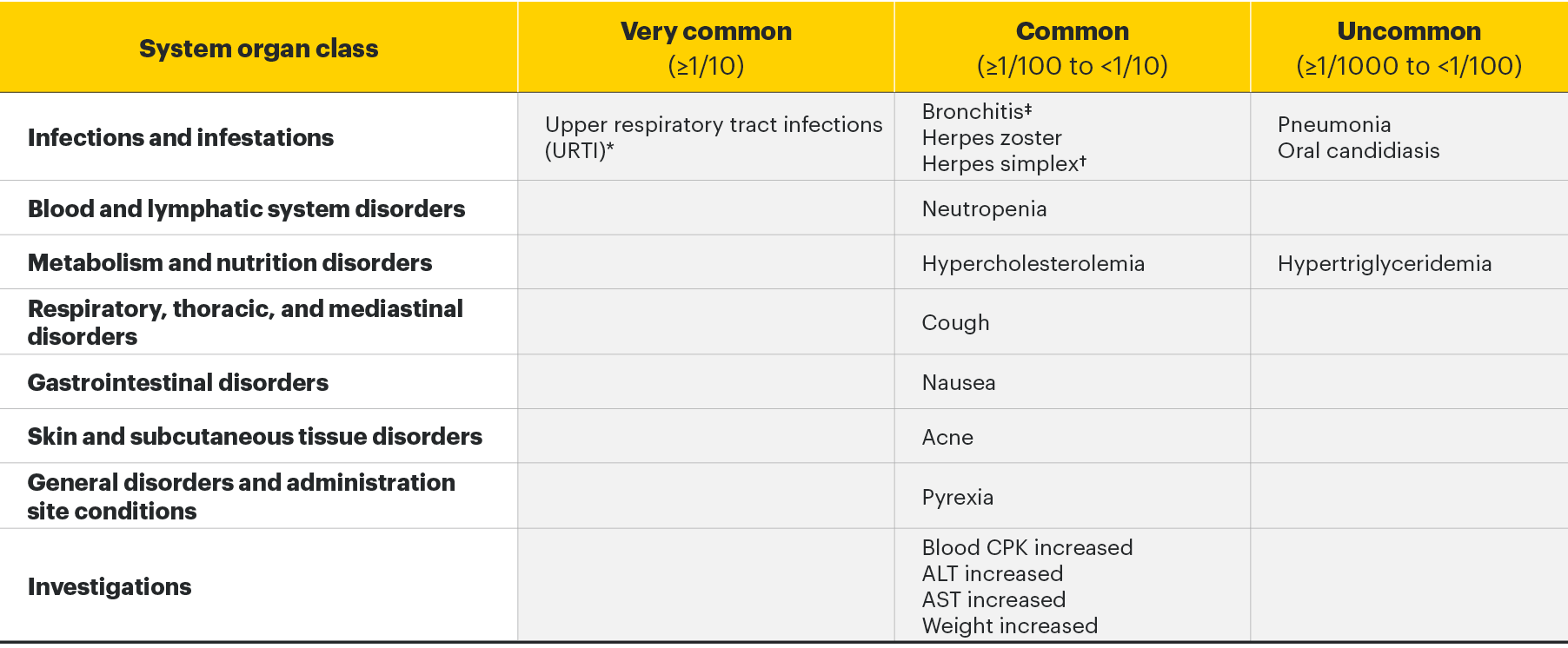
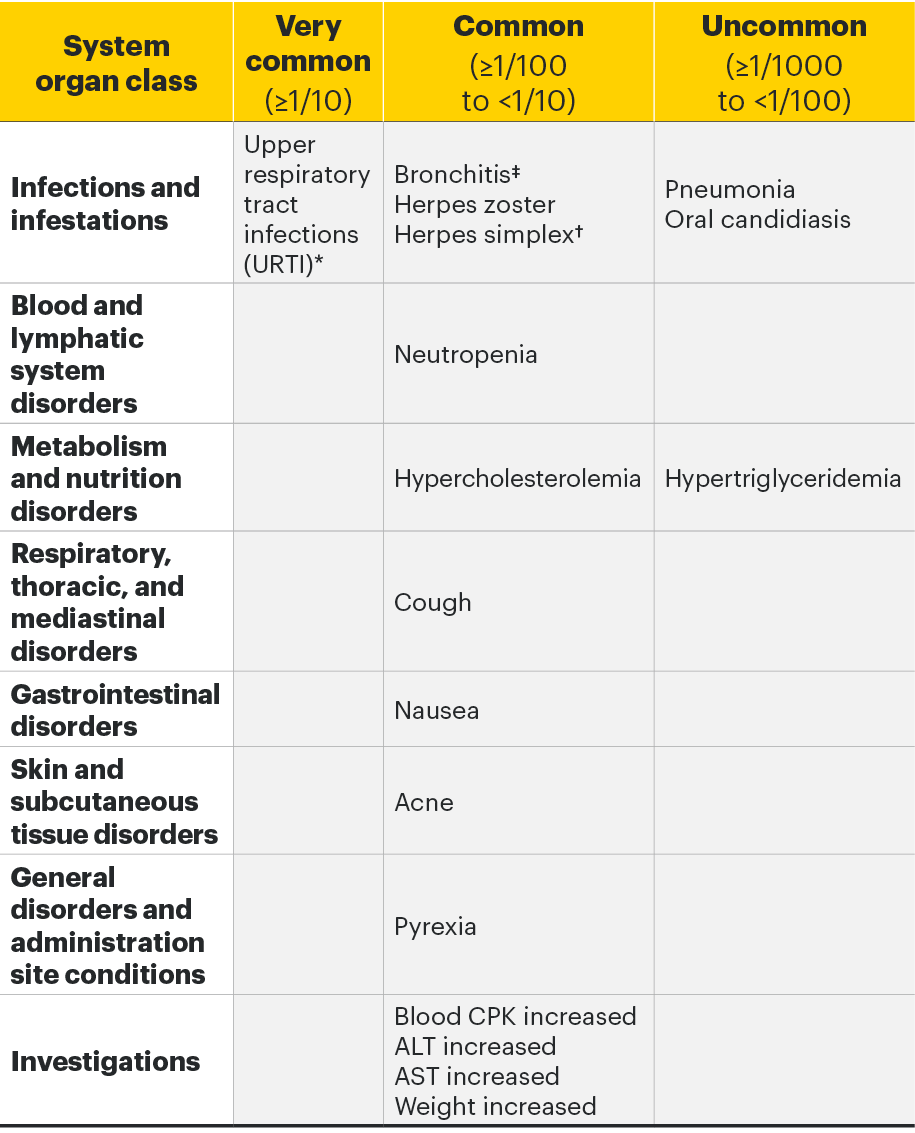
*Includes upper respiratory tract infection, acute sinusitis, laryngitis, nasopharyngitis, oropharyngeal pain, pharyngitis, pharyngotonsillitis, rhinitis, sinusitis, tonsillitis, viral upper respiratory tract infection.
†Includes herpes simplex and oral herpes.
‡Includes bronchitis, bronchitis viral, bronchitis bacterial, and tracheobronchitis.
For more detailed safety information, please see the Important Safety Information link at the top of the page, or for complete prescribing information, the RINVOQ SmPC.
Special Warnings and Precautions3
- Serious infections: RINVOQ should not be initiated in patients with an active, serious infection, including localized infections. Closely monitor patients for the development of signs and symptoms of infection during and after treatment with RINVOQ. If a patient develops a serious or opportunistic infection, RINVOQ should be interrupted. As there is a higher incidence of infections in the elderly ≥65 years of age, caution should be used when treating this population.
- Tuberculosis: Patients should be screened for tuberculosis (TB) before starting RINVOQ. Treatment should not be initiated in patients with active TB. Anti-TB therapy should be considered prior to initiation of RINVOQ in patients with previously untreated latent TB or in patients with risk factors for TB infection. Monitor patients for the development of signs and symptoms of TB, including patients who tested negative for latent TB infection prior to initiating therapy.
- Viral reactivation: Screening for viral hepatitis and monitoring for reactivation should be performed before starting and during treatment with RINVOQ. The risk of herpes zoster appears to be higher in Japanese patients treated with upadacitinib. If a patient develops herpes zoster, consider interruption of RINVOQ therapy until the episode resolves. If hepatitis B virus DNA is detected while receiving RINVOQ, a liver specialist should be consulted.
- Nonmelanoma skin cancer: Periodic skin examination is recommended for patients who are at increased risk for skin cancer.
- Vaccinations: Use of live, attenuated vaccines during or immediately prior to RINVOQ treatment is not recommended. Prior to initiating RINVOQ, it is recommended that patients be brought up to date with all immunizations, including prophylactic zoster vaccinations, in agreement with current immunization guidelines.
- Venous thromboembolism: RINVOQ should be used with caution in patients at high risk for deep venous thrombosis (DVT)/pulmonary embolism (PE). If clinical features of DVT/PE occur, RINVOQ treatment should be discontinued and patients should be evaluated promptly, followed by appropriate treatment.
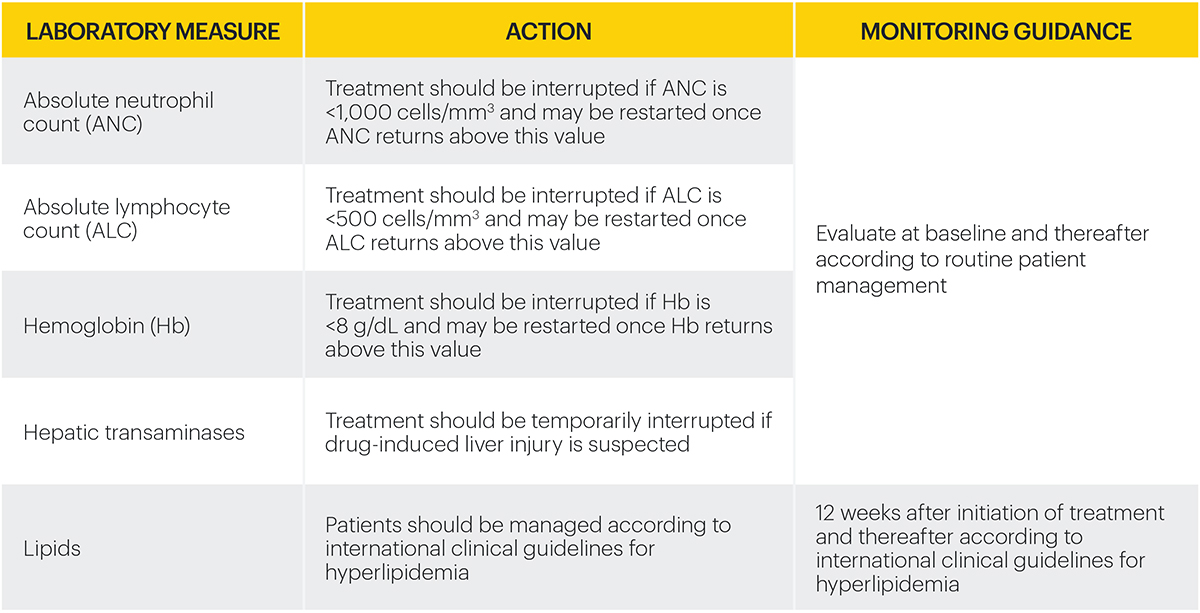
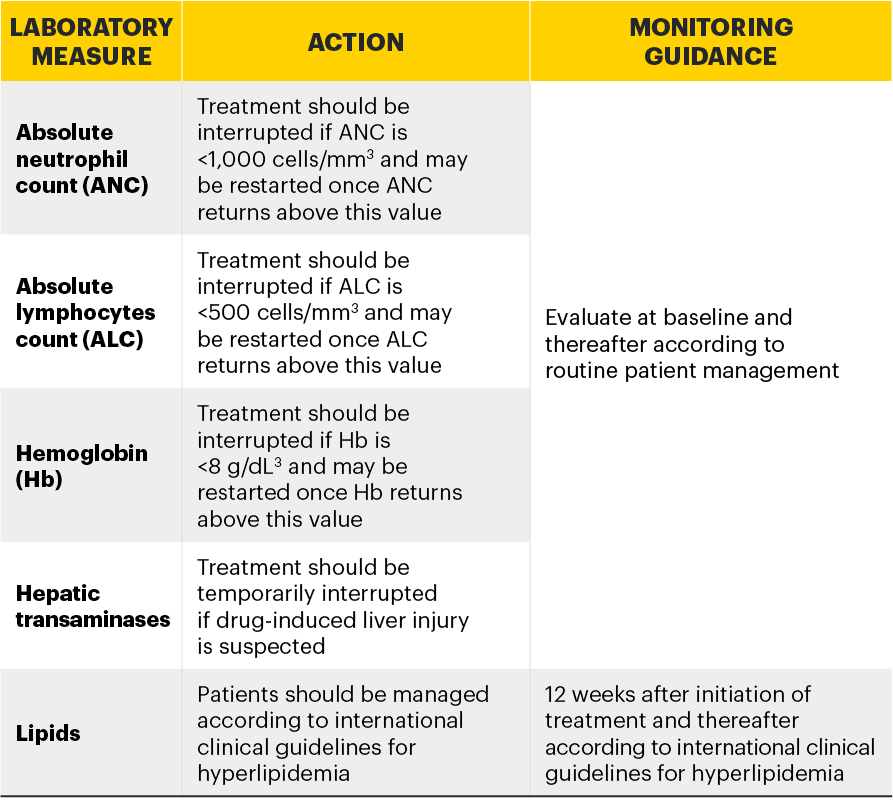
Monitoring requirements
Treatment with RINVOQ should not be initiated if:3
- Absolute neutrophil count <1,000 cells/mm3*
- Absolute lymphocyte count <500 cells/mm3*
- Hemoglobin <8 g/dL*
Laboratory measures and monitoring guidance3
Elderly
No dose adjustment is required in patients aged 65 years and older. There are limited data in patients aged 75 years and older.
Renal impairment
No dose adjustment is required in patients with mild or moderate renal impairment. There are limited data on the use of RINVOQ in subjects with severe renal impairment. RINVOQ should be used with caution in patients with severe renal impairment. The use of upadacitinib has not been studied in subjects with end-stage renal disease.2
Hepatic impairment
No dose adjustment is required in patients with mild (Child Pugh A) or moderate (Child Pugh B) hepatic impairment. RINVOQ should not be used in patients with severe (Child Pugh C) hepatic impairment.2
Pediatric
The safety and efficacy of RINVOQ in children and adolescents aged 0 to less than 18 years have not yet been established. No data are available.
*Treatment may be initiated or restarted after levels return above specified values.

Safety information3
RINVOQ is contraindicated in patients hypersensitive to the active substance or to any of the excipients, in patients with active tuberculosis (TB) or active serious infections, in patients with severe hepatic impairment, and during pregnancy.
Use in combination with other potent immunosuppressants is not recommended.
Serious and sometimes fatal infections have been reported in patients receiving upadacitinib. The most frequent serious infections reported included pneumonia and cellulitis. Cases of bacterial meningitis have been reported. Among opportunistic infections, TB, multidermatomal herpes zoster, oral/esophageal candidiasis, and cryptococcosis have been reported with upadacitinib. Prior to initiating upadacitinib, consider the risks and benefits of treatment in patients with chronic or recurrent infection or with a history of a serious or opportunistic infection, in patients who have been exposed to TB or have resided or traveled in areas of endemic TB or endemic mycoses, and in patients with underlying conditions that may predispose them to infection. Upadacitinib therapy should be interrupted if a patient develops a serious or opportunistic infection. As there is a higher incidence of infections in patients ≥65 years of age, caution should be used when treating this population.
Patients should be screened for TB before starting upadacitinib therapy. Anti-TB therapy should be considered prior to initiation of upadacitinib in patients with previously untreated latent TB or in patients with risk factors for TB infection.
Viral reactivation, including cases of herpes zoster, were reported in clinical studies. The risk of herpes zoster appears to be higher in Japanese patients treated with upadacitinib. Consider interruption of therapy if a patient develops herpes zoster until the episode resolves. Screening for viral hepatitis and monitoring for reactivation should be performed before starting and during therapy with upadacitinib.
The use of live, attenuated vaccines during or immediately prior to therapy is not recommended. It is recommended that patients be brought up to date with all immunizations, including prophylactic zoster vaccinations, prior to initiating upadacitinib, in agreement with current immunization guidelines.
The risk of malignancies, including lymphoma is increased in patients with rheumatoid arthritis (RA). Immunomodulatory medicinal products may increase the risk of malignancies, including lymphoma. The clinical data are currently limited and long-term studies are ongoing. Malignancies, including nonmelanoma skin cancer (NMSC), have been reported in patients treated with upadacitinib. Consider the risks and benefits of upadacitinib treatment prior to initiating therapy in patients with a known malignancy other than a successfully treated NMSC or when considering continuing upadacitinib therapy in patients who develop a malignancy. Periodic skin examination is recommended for patients who are at increased risk for skin cancer.
Absolute neutrophil count <1000 cells/mm3, absolute lymphocyte count <500 cells/mm3, or hemoglobin levels <8 g/dL were reported in ≤1% of patients in clinical trials. Treatment should not be initiated, or should be temporarily interrupted, in patients with these hematological abnormalities observed during routine patient management.
RA patients have an increased risk for cardiovascular disorders. Patients treated with upadacitinib should have risk factors (e.g., hypertension, hyperlipidemia) managed as part of usual standard of care.
Upadacitinib treatment was associated with increases in lipid parameters, including total cholesterol, low-density lipoprotein cholesterol, and high-density lipoprotein cholesterol. The effect of these lipid parameter elevations on cardiovascular morbidity and mortality has not been determined.
Treatment with upadacitinib was associated with an increased incidence of liver enzyme elevation compared to placebo. If increases in ALT or AST are observed during routine patient management and drug-induced liver injury is suspected, upadacitinib therapy should be interrupted until this diagnosis is excluded.
Events of deep vein thrombosis (DVT) and pulmonary embolism (PE) have been reported in patients receiving JAK inhibitors, including upadacitinib. Upadacitinib should be used with caution in patients at high risk for DVT/PE. Risk factors that should be considered in determining the patient’s risk for DVT/PE include older age, obesity, a medical history of DVT/PE, patients undergoing major surgery, and prolonged immobilization. If clinical features of DVT/PE occur, upadacitinib treatment should be discontinued and patients should be evaluated promptly, followed by appropriate treatment.
The most commonly reported adverse drug reactions (ADRs) were upper respiratory tract infections, bronchitis, nausea, blood creatine phosphokinase (CPK) increased, and cough. The most common serious adverse reactions were serious infections.
Please see the RINVOQ Summary of Product Characteristics for complete Prescribing Information.


I want to receive more information via a product specialist.
I have a question for Medical Information.
References
- Cohen SB, van Vollenhoven RF, Winthrop KL, et al. Safety profile of upadacitinib in rheumatoid arthritis: integrated analysis from the SELECT phase III clinical programme. Ann Rheum Dis. 2020;80(3):304-311. doi:10.1136/annrheumdis-2020-218510
- Cohen SB, van Vollenhoven R, Curtis JR, et al. Safety profile of upadacitinib up to 3 years of exposure in patients with rheumatoid arthritis. Poster presented at: European E-Congress of Rheumatology (EULAR); June 3–6, 2020.
- RINVOQ [Summary of Product Characteristics]. AbbVie Deutschland GmbH & Co. KG; May 2021.